
1. Einführung
Zuletzt aktualisiert: 25.04.2022
Überblick
In diesem Codelab stellen Sie einen automatisch skalierenden High Performance Computing-Cluster (HPC) in Google Cloud bereit.Dieser Cluster wird mit einer Terraform-Bereitstellung erstellt und Gromacs wird über Spack installiert. Der Cluster wird mit dem Slurm-Jobplaner verwaltet. Nachdem der Cluster erstellt wurde, führen Sie die benchMEM-, benchPEP- oder benchRIB-Benchmarks aus.
Lerninhalte
- So stellen Sie einen HPC-Cluster bereit, der den Slurm-Job-Scheduler unterstützt
- So führen Sie GPU-beschleunigte Molekulardynamik-Simulationen mit GROMACS in Google Cloud mit einem Slurm-Batchjob aus
Das brauchen Sie
2. Einrichtung
Für dieses Codelab müssen Sie die Compute Engine API und die Cloud Build API aktivieren. Öffnen Sie Cloud Shell und führen Sie die folgenden Befehle aus, um die Compute Engine API und die Cloud Build API zu aktivieren. Denken Sie daran,
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Achtung:Wenn Sie planen, eine Verbindung zu Ihrem Cluster über Drittanbieter-SSH (z.B. OpenSSH) herzustellen, müssen Sie Ihrem Cloud Identity-Profil mit OS Login einen SSH-Schlüssel anhängen. Weitere Informationen zum Hinzufügen von SSH-Schlüsseln zu Ihrem Cloud Identity-Profil
3. [OPTIONAL] GCP-VM-Image mit Gromacs erstellen
Für dieses Codelab haben wir ein vorgefertigtes Image bereitgestellt, da die Installation von Gromacs und allen zugehörigen Abhängigkeiten bis zu zwei Stunden dauern kann. Wenn Sie dieses vorgefertigte Image verwenden möchten, um Zeit zu sparen, können Sie mit dem nächsten Abschnitt fortfahren.
Wenn Sie Forschungsanwendungen in Google Cloud ausführen, haben Sie viele Möglichkeiten, Ihre Anwendung zu installieren und bereitzustellen. In diesem Abschnitt des Codelabs erstellen Sie ein VM-Image, das auf dem VM-Image slurm-gcp (CentOS7) basiert. Während des Erstellungsprozesses werden der Compiler, alle Abhängigkeiten von Gromacs und Gromacs installiert.
Die Gromacs Cloud Build-Pipeline im RCC Apps-Repository enthält die erforderlichen Anleitungen für die Installation von Gromacs. Beim Installationsprozess wird Packer verwendet, um eine VM bereitzustellen, auf der Spack installiert wird. Dadurch werden der GCC@9.2.0-Compiler und Gromacs@2021.2 mit aktivierter GPU-Beschleunigung installiert.
- Öffnen Sie Cloud Shell in der GCP.
- Repository „FluidNumerics/rcc-apps“ klonen
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Erstellen Sie das Image mit Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Den Status Ihres Build-Prozesses können Sie im Google Cloud Build-Dashboard prüfen.
Der Build-Prozess kann bis zu zwei Stunden dauern. Um dies zu beschleunigen, können Sie das Schema Ihrer Build-Konfigurationsdatei ändern, um den Maschinentyp zu ändern und so die Build-Leistung zu verbessern. Dazu können Sie die Build-Variable _MACHINE_TYPE verwenden. Beispiel:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Nachdem der Build abgeschlossen ist, steht in Ihrem Google Cloud-Projekt ein VM-Image zur Verfügung, mit dem Sie Ihren Cluster bereitstellen können.
4. Automatisch skalierenden HPC-Cluster mit Terraform bereitstellen
In diesem Abschnitt stellen Sie mit Terraform einen automatisch skalierenden HPC-Cluster mit installiertem Slurm-Job-Scheduler bereit. Dieser Cluster wird mit Rechenknoten bereitgestellt, die jeweils 8 vCPUs und 1 Nvidia® Tesla V100-GPU haben.
- Öffnen Sie Cloud Shell in der GCP.
- Repository „FluidNumerics/rcc-apps“ klonen
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Wechseln Sie in das GROMACS-Terraform-Verzeichnis:
cd ~/rcc-apps/gromacs/tf/slurm
- Terraform-Plan erstellen und prüfen Legen Sie die Umgebungsvariablen
GMX_NAME,GMX_PROJECTundGMX_ZONEfest, um den Namen Ihres Clusters, Ihr GCP-Projekt und die Zone anzugeben, in der Sie die Bereitstellung vornehmen möchten. Wenn Sie sich nicht sicher sind, lesen Sie bitte den Hinweis unten.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Wenn Sie im vorherigen Abschnitt dieses Codelabs ein eigenes VM-Image erstellt haben, müssen Sie auch die Umgebungsvariable GMX_IMAGE festlegen.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Erstellen Sie den Plan mit dem Befehl „make“, der
terraform init && terraform planausführt.
make plan
- Stellen Sie den Cluster bereit. Die Einrichtung dauert nur wenige Minuten, da Gromacs und seine Abhängigkeiten auf Ihrem Cluster vorinstalliert sind.
make apply
- Stellen Sie eine SSH-Verbindung zum Anmeldeknoten her, der im vorherigen Schritt erstellt wurde. Sie können diesen Knoten im vorherigen Schritt sehen (wahrscheinlich gromacs-login0). Klicken Sie dazu im Konsolenmenüelement Compute Engine -> VM-Instanz neben der Liste der VM-Instanzen auf die Schaltfläche „SSH“.
Option:Mit diesem Paar von gcloud-Befehlen wird der Name des Anmeldeknotens ermittelt und eine SSH-Verbindung zu ihm hergestellt:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Nachdem Sie eine Verbindung zum Anmeldeknoten hergestellt haben, prüfen Sie, ob Gromacs installiert ist, um die Einrichtung des Clusters zu bestätigen.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Prüfen Sie, ob
/opt/share/gromacsdie unten aufgeführten Inhalte enthält.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. benchRIB-Benchmark ausführen
Gromacs ist eine Forschungssoftware, mit der die Molekulardynamik simuliert und Molekularstrukturen unter Berücksichtigung von Energieeinsparungen berechnet werden können. Die Benchmarks, die im VM-Image für dieses Codelab enthalten sind, konzentrieren sich auf die Molekulardynamik, die Entwicklung von Atomsystemen.
In der Molekulardynamik werden die Positionen, Geschwindigkeiten und Beschleunigungen von Atomen mithilfe der Newtonschen Bewegungsgesetze simuliert :
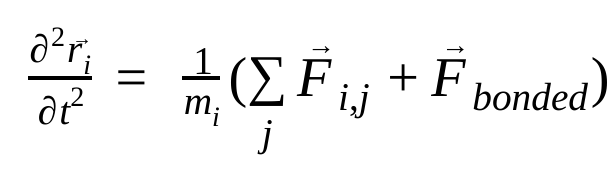
Dabei ist 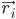 die Position des Atoms i, t die Zeit,
die Position des Atoms i, t die Zeit, 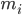 die Masse des Atoms i und
die Masse des Atoms i und 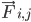 die nicht gebundene Kraft auf das Atom i aufgrund des Atoms j und
die nicht gebundene Kraft auf das Atom i aufgrund des Atoms j und 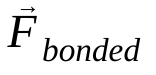 die Kräfte aufgrund gebundener Wechselwirkungen. Anhand der Temperatur, des Drucks, der Atompositionen und der Atomgeschwindigkeiten werden die Kräfte berechnet und das System wird numerisch integriert, um neue Atomgeschwindigkeiten und -positionen zu erhalten. Dieser Vorgang wird wiederholt, um die Molekulardynamik für einen bestimmten Zeitraum zu simulieren.
die Kräfte aufgrund gebundener Wechselwirkungen. Anhand der Temperatur, des Drucks, der Atompositionen und der Atomgeschwindigkeiten werden die Kräfte berechnet und das System wird numerisch integriert, um neue Atomgeschwindigkeiten und -positionen zu erhalten. Dieser Vorgang wird wiederholt, um die Molekulardynamik für einen bestimmten Zeitraum zu simulieren.
Das Gromacs-Image (entweder das von Ihnen erstellte oder das bereitgestellte) enthält drei Benchmarks.
- benchMEM
- benchRIB
- benchPEP
Diese Benchmarks stammen aus Dr. Kutzner's set of Free Gromacs Benchmarks und sind ein Standardsatz von transienten Molekulardynamiksimulationen. Die Anzahl der Atome und die Dauer der Simulation variieren je nach Benchmark. Die relevanten Konfigurationen für jede Simulation sind in der Tabelle unten aufgeführt.
Messwert / Benchmark | benchMEM | benchRIB | benchPEP |
Anzahl der Atome | 81.743 | 2.136.412 | 12.495.503 |
Systemgröße / nm | 10,8 × 10,2 × 9,6 | 31,2 × 31,2 × 31,2 | 50,0 × 50,0 × 50,0 |
Zeitschritt / fs | 2 | 4 | 2 |
Grenzradien / nm | 1 | 1 | 1.2 |
PME-Rasterabstand / nm | 0,12 | 0,135 | 0,16 |
Um den Benchmark auszuführen, senden Sie einen Slurm-Batchjob. Standardmäßig wird mit dem bereitgestellten Batch-Skript der benchRIB-Benchmark ausgeführt. Die Eingabe-Decks, die die Konfigurationen für die bereitgestellten Benchmarks definieren, sind im Gromacs-VM-Image unter /opt/share/gromacs enthalten. Außerdem ist unter /opt/share ein Beispiel für ein Bash-Skript zum Ausführen von Gromacs verfügbar.
Für diesen Abschnitt müssen Sie eine SSH-Verbindung zum Anmeldeknoten des Clusters herstellen.
- Batchjob mit dem Befehl „sbatch“ senden
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Dadurch wird der Job zur Ausführung in die Warteschlange gestellt und Slurm stellt einen Compute-Knoten für Sie bereit. Wenn Sie sinfo ausführen, sehen Sie, dass sich ein Rechenknoten im Status alloc# befindet. Das bedeutet, dass der Rechenknoten für Ihren Job zugewiesen wurde, aber gerade bereitgestellt wird. Sobald Ihr Job ausgeführt wird, wird der Knoten auf den Status alloc gesetzt.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Warten Sie, bis der Job abgeschlossen ist. Der Standard-Benchmark (benchRIB) simuliert etwa 8 Millionen Atome und ist für 5.000 Zeitschritte (mit 4 Zeitschritten/fs) konfiguriert. Die Ausführung dauert etwa 6 Minuten. Sie können den Status Ihres Jobs mit diesem Befehl überwachen:
watch squeue
Wenn Ihr Job aus der Warteschlange entfernt wurde, können Sie mit Strg + C beenden.
- Wenn der Job abgeschlossen ist, sollte in Ihrem aktuellen Verzeichnis ein Verzeichnis namens
run/angezeigt werden, das die Simulationsausgabe (unterrun/MEM) und eine Logdatei namensgromacs.outenthält. Das Verzeichnisrun/MEMenthält die beiden Dateienener.edrundmd.log. In der Dateiener.edrwerden die Energien, die Temperatur, der Druck, die Boxgröße, die Dichte und die Virials des Systems in einem portablen Binärformat gespeichert. Wie der Name der Erweiterung schon sagt, enthält die Dateimd.logLogs für die Gromacs-Simulation. Neben den Logging-Informationen der Particle-Particle- und PME-Solver sind auch Informationen zur Simulationsleistung enthalten. Der Inhalt von „gromacs.out“ enthält die Standardausgabe vongmx mdrunund sollte in etwa so aussehen:
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Glückwunsch
In diesem Codelab haben Sie einen automatisch skalierenden, cloudnativen HPC-Cluster erstellt und eine GPU-beschleunigte Molekulardynamiksimulation mit Gromacs in Google Cloud ausgeführt.
Bereinigen
So vermeiden Sie, dass Ihrem Google Cloud-Konto die in diesem Codelab verwendeten Ressourcen in Rechnung gestellt werden:
EMPFOHLEN:HPC-Cluster mit Terraform löschen
- Öffnen Sie Cloud Shell und wechseln Sie zum Beispielverzeichnis
gromacs/tf/slurm.
cd ~/rcc-apps/gromacs/tf/slurm
- Führen Sie „make destroy“ aus, um alle Ressourcen zu löschen.
make destroy
ODER: Projekt löschen (am effektivsten und zerstörerischsten)
Am einfachsten vermeiden Sie weitere Kosten, indem Sie das für das Codelab erstellte Projekt löschen.
Achtung: Das Löschen von Projekten hat folgende Auswirkungen:
- Alle Inhalte des Projekts werden gelöscht. Wenn Sie für dieses Codelab ein bereits bestehendes Projekt verwendet haben, werden auch alle anderen im Rahmen des Projekts erstellten Daten gelöscht.
- Benutzerdefinierte Projekt-IDs gehen verloren. Beim Erstellen dieses Projekts haben Sie möglicherweise eine benutzerdefinierte Projekt-ID erstellt, die Sie weiterhin verwenden möchten. Damit die URLs, die die Projekt-ID verwenden, z. B. eine appspot.com-URL, erhalten bleiben, sollten Sie ausgewählte Ressourcen innerhalb des Projekts löschen, statt das gesamte Projekt.
Wenn Sie mehrere Codelabs und Kurzanleitungen durcharbeiten möchten, können Sie die Überschreitung von Projektkontingenten verhindern, indem Sie Projekte wiederverwenden.
- Wechseln Sie in der Cloud Console zur Seite Ressourcen verwalten. Zur Seite „Ressourcen verwalten“
- Wählen Sie in der Projektliste das Projekt aus, das Sie löschen möchten, und klicken Sie dann auf Löschen
 .
. - Geben Sie im Dialogfeld die Projekt-ID ein und klicken Sie auf Beenden, um das Projekt zu löschen.