
1. מבוא
עדכון אחרון: 25 באפריל 2022
מה תפַתחו
ב-codelab הזה תפרסו אשכול מחשוב בעל ביצועים גבוהים (HPC) עם התאמה אוטומטית לעומס ב-Google Cloud.פריסת Terraform יוצרת את האשכול הזה עם Gromacs שהותקן באמצעות Spack. האשכול ינוהל באמצעות מתזמן המשימות Slurm. אחרי שיוצרים את האשכול, מריצים את מדדי הביצועים benchMEM, benchPEP או benchRIB.
מה תלמדו
- איך פורסים אשכול HPC שתומך במתזמן המשימות Slurm
- איך מריצים סימולציות של דינמיקה מולקולרית עם האצת GPU באמצעות Gromacs ב-Google Cloud באמצעות משימת אצווה של Slurm
מה צריך
2. הגדרה
כדי להשלים את ה-codelab הזה , צריך להפעיל את ממשקי ה-API של Compute Engine ו-Cloud Build. כדי להפעיל את Compute Engine API ואת Cloud Build API, פותחים את Cloud Shell ומריצים את הפקודות הבאות. חשוב לזכור להחליף את project-id ב-
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
זהירות: אם אתם מתכננים להשתמש ב-SSH של צד שלישי (למשל OpenSSH) כדי להתחבר לאשכול, הקפידו לצרף מפתח SSH לפרופיל הזהות בענן באמצעות OS Login. מידע נוסף על הוספת מפתחות SSH לפרופיל Cloud Identity
3. [אופציונלי] יצירת קובץ אימג' של מכונה וירטואלית ב-GCP באמצעות Gromacs
ב-codelab הזה, סיפקנו לכם תמונה מוכנה מראש, כי תהליך ה-build יכול להימשך עד שעתיים עד להתקנת Gromacs וכל התלות שלו. אם אתם רוצים להשתמש בתמונה המוכנה מראש הזו כדי לחסוך זמן, אתם יכולים לדלג לסעיף הבא.
כשמריצים אפליקציות מחקר ב-Google Cloud, יש הרבה אפשרויות להתקנה ולפריסה של האפליקציה. בקטע הזה של ה-Codelab, תיצרו תמונה של מכונה וירטואלית שמבוססת על תמונה של מכונה וירטואלית מסוג slurm-gcp (CentOS7). במהלך תהליך היצירה, המהדר, כל יחסי התלות של Gromacs ו-Gromacs יותקנו.
פייפליין ה-Build של Gromacs Cloud במאגר האפליקציות של RCC כולל את ההוראות הנדרשות להתקנת Gromacs. בתהליך ההתקנה נעשה שימוש ב-Packer כדי לפרוס מכונה וירטואלית שמתקינה את Spack, שבתורה מתקינה את קומפיילר GCC@9.2.0 ואת Gromacs@2021.2 עם הפעלת האצת GPU.
- פותחים את Cloud Shell ב-GCP.
- משכפלים את המאגר FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- יוצרים את קובץ האימג' באמצעות Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
אפשר לבדוק את הסטטוס של תהליך ה-Build בלוח הבקרה של Google Cloud Build.
תהליך ה-build עשוי להימשך עד שעתיים. כדי להאיץ את התהליך,כדאי לשנות את סכימת קובץ התצורה של ה-build כדי לשנות את סוג המכונה ולשפר את ביצועי ה-build. אפשר לעשות זאת באמצעות משתנה ה-build _MACHINE_TYPE. לדוגמה:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
אחרי שהבנייה תסתיים, תהיה לכם תמונה של מכונה וירטואלית בפרויקט בענן שלכם ב-Google Cloud, שתוכלו להשתמש בה כדי לפרוס את האשכול.
4. פריסת אשכול HPC עם התאמה אוטומטית לעומס באמצעות Terraform
בקטע הזה תשתמשו ב-Terraform כדי לפרוס אשכול HPC עם התאמה אוטומטית לעומס, שבו מותקן מתזמן המשימות Slurm. האשכול הזה יופעל עם צמתי מחשוב שלכל אחד מהם יש 8 מעבדי CPU וירטואליים ומעבד GPU אחד מסוג Nvidia® Tesla V100.
- פותחים את Cloud Shell ב-GCP.
- משכפלים את המאגר FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- עוברים לספריית gromacs terraform:
cd ~/rcc-apps/gromacs/tf/slurm
- יוצרים תוכנית Terraform ובודקים אותה. מגדירים את משתני הסביבה
GMX_NAME,GMX_PROJECTו-GMX_ZONEכדי לציין את השם של האשכול, את פרויקט GCP ואת האזור שבו רוצים לבצע פריסה. אם אתם לא בטוחים, כדאי לעיין בהערה שלמטה
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- אם יצרתם תמונת מכונה וירטואלית משלכם בחלק הקודם של ה-Codelab הזה, תצטרכו גם להגדיר את משתנה הסביבה GMX_IMAGE.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- יוצרים את התוכנית באמצעות הפקודה make, שתריץ את
terraform init && terraform plan.
make plan
- פורסים את האשכול. תהליך ההגדרה נמשך רק כמה דקות כי Gromacs והתלויות שלו מותקנים מראש באשכול.
make apply
- מתחברים באמצעות SSH לצומת הכניסה שנוצר בשלב הקודם. אפשר לראות את הצומת הזה בשלב הקודם (כנראה בשם gromacs-login0). כדי לעשות זאת, לוחצים על הלחצן SSH שליד רשימת מכונות ה-VM בפריט התפריט Compute Engine -> VM instance במסוף.
אפשרות: צמד הפקודות הבא של gcloud יזהה את שם צומת הכניסה ויבצע SSH אליו:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- אחרי שמתחברים לצומת הכניסה, כדי לאמת את הגדרת האשכול, בודקים ש-Gromacs מותקן.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- צריך לוודא שהתוכן של
/opt/share/gromacsהוא כמו שמופיע בהמשך.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. הפעלת ההשוואה לשוק benchRIB
Gromacs היא תוכנת מחקר שמשמשת לסימולציה של דינמיקה מולקולרית ולחישוב מבנים מולקולריים במסגרת אילוצים של מזעור אנרגיה. המדדים שמופיעים בתמונת מכונת ה-VM ב-Codelab הזה מתמקדים בדינמיקה מולקולרית, כלומר בהתפתחות של מערכות אטומים.
בדינמיקה מולקולרית, המיקומים, המהירויות והתאוצות של האטומים מודמים באמצעות חוקי התנועה של ניוטון :
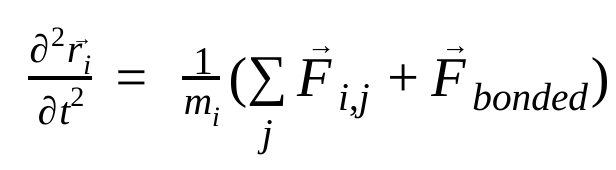
כאשר 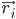 הוא המיקום של אטום i, t הוא הזמן,
הוא המיקום של אטום i, t הוא הזמן, 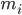 הוא המסה של אטום i,
הוא המסה של אטום i, 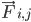 הוא הכוח הלא קוולנטי על אטום i בגלל אטום j, ו-
הוא הכוח הלא קוולנטי על אטום i בגלל אטום j, ו-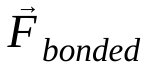 הם הכוחות בגלל אינטראקציות קוולנטיות. בהינתן הטמפרטורה, הלחץ, מיקומי האטומים והמהירויות שלהם, הכוחות מחושבים והמערכת משולבת באופן מספרי כדי לקבל מהירויות ומיקומים חדשים של האטומים. התהליך הזה חוזר על עצמו כדי לדמות את הדינמיקה המולקולרית למשך תקופה מסוימת.
הם הכוחות בגלל אינטראקציות קוולנטיות. בהינתן הטמפרטורה, הלחץ, מיקומי האטומים והמהירויות שלהם, הכוחות מחושבים והמערכת משולבת באופן מספרי כדי לקבל מהירויות ומיקומים חדשים של האטומים. התהליך הזה חוזר על עצמו כדי לדמות את הדינמיקה המולקולרית למשך תקופה מסוימת.
תמונת Gromacs (זו שיצרתם או זו שסופקה) מגיעה עם שלושה מדדים
- benchMEM
- benchRIB
- benchPEP
הנקודות להשוואה האלה מגיעות מקבוצת הנקודות להשוואה של Gromacs בחינם של ד"ר קוצנר והן קבוצה סטנדרטית של סימולציות דינמיקה מולקולרית חולפת. כל נקודת השוואה שונה במספר האטומים ובמשך הסימולציה. ההגדרות הרלוונטיות לכל סימולציה מפורטות בטבלה הבאה.
מדד / נקודת השוואה | benchMEM | benchRIB | benchPEP |
מספר האטומים | 81,743 | 2,136,412 | 12,495,503 |
גודל המערכת / nm | 10.8 x 10.2 x 9.6 | 31.2 x 31.2 x 31.2 | 50.0 x 50.0 x 50.0 |
Time Step / fs | 2 | 4 | 2 |
רדיוסים של מועד אחרון להזמנה / מייל ימי | 1 | 1 | 1.2 |
PME grid spacing / nm | 0.12 | 0.135 | 0.16 |
כדי להריץ את ההשוואה, שולחים משימה באצווה של Slurm. כברירת מחדל, סקריפט האצווה שסופק מריץ את מדד הביצועים benchRIB. חבילות הקלט שמגדירות את התצורות עבור המדדים שסופקו כלולות בתמונת המכונה הווירטואלית של Gromacs בתיקייה /opt/share/gromacs. בנוסף, סקריפט Bash לדוגמה להרצת Gromacs זמין ב-/opt/share.
בקטע הזה, צריך להשתמש ב-SSH כדי להתחבר לצומת הכניסה של האשכול.
- שליחת משימה באצווה באמצעות הפקודה sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
הפעולה הזו תכניס את העבודה לתור לביצוע, ו-Slurm יקצה לכם צומת חישוב. כשמריצים את הפקודה sinfo, רואים שצומת החישוב נמצא במצב alloc#, כלומר צומת החישוב הוקצה לעבודה שלכם, אבל הוא בתהליך הקצאה. אחרי שהעבודה תפעל, הצומת יוגדר למצב alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
ממתינים לסיום העבודה. המדד שמוגדר כברירת מחדל (benchRIB) מדמה כ-8 מיליון אטומים, והוא מוגדר להרצה של 5,000 צעדים בזמן (עם 4 צעדים בזמן לכל fs) ולוקח כ-6 דקות להשלמה. אפשר לעקוב אחרי הסטטוס של העבודה באמצעות הפקודה הבאה:
watch squeue
כשהעבודה שלכם נעלמת מהתור, אפשר לצאת באמצעות Ctrl+C.
- בסיום העבודה, אמורה להופיע בספרייה הנוכחית ספרייה בשם
run/שמכילה את פלט הסימולציה (בקטעrun/MEM) וקובץ יומן בשםgromacs.out. הספרייהrun/MEMמכילה שני קבצים:ener.edrו-md.log. בקובץener.edrמאוחסנים נתוני האנרגיה, הטמפרטורה, הלחץ, גודל התיבה, הצפיפות והוויראליות של המערכת בפורמט בינארי נייד. כפי שאפשר להבין מהשם של התוסף, קובץmd.logמכיל יומנים של סימולציית Gromacs וכולל מידע על ביצועי הסימולציה, בנוסף למידע מהיומנים של פותרי הבעיות של חלקיקים ו-PME. התוכן של gromacs.out מכיל פלט רגיל מ-gmx mdrunוצריך להיראות בערך כמו מה שמוצג למטה.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. מזל טוב
ב-codelab הזה יצרתם אשכול HPC מותאם לענן עם התאמה אוטומטית לעומס, והפעלתם סימולציה של דינמיקה מולקולרית עם האצת GPU באמצעות Gromacs ב-Google Cloud.
סידור וארגון
כדי להימנע מחיובים בחשבון Google Cloud על המשאבים שבהם השתמשתם ב-codelab הזה:
מומלץ: מחיקת אשכול HPC באמצעות Terraform
- פותחים את Cloud Shell ועוברים לספריית הדוגמאות
gromacs/tf/slurm.
cd ~/rcc-apps/gromacs/tf/slurm
- מריצים את הפקודה make destroy כדי למחוק את כל המשאבים.
make destroy
או, מחיקת הפרויקט (הפעולה הכי יעילה והכי הרסנית)
הדרך הקלה ביותר לבטל את החיוב היא למחוק את הפרויקט שיצרתם בשביל ה-Codelab.
זהירות: כשמוחקים פרויקט:
- התוכן של הפרויקט נמחק כולו. אם השתמשתם בפרויקט קיים כדי לתרגל את ה-Codelab הזה, כשתמחקו אותו תימחק גם כל העבודה שביצעתם באותו פרויקט.
- מאבדים את מזהה הפרויקט בהתאמה אישית. כשיצרתם את הפרויקט, יכולתם לבחור לו מזהה פרויקט בהתאמה אישית כדי להשתמש בו בעתיד. כדי לשמור על כתובות ה-URL שמשתמשות במזהה הפרויקט, כמו כתובת URL של appspot.com, צריך למחוק את המשאבים שנבחרו בפרויקט אבל לא את הפרויקט כולו.
אם אתם מתכננים להיעזר בכמה הדרכות של Codelab או מדריכים למתחילים, מומלץ להשתמש שוב באותו פרויקט כדי לא לחרוג ממכסות הפרויקטים.
- במסוף Cloud, נכנסים לדף Manage resources. כניסה לדף Manage resources
- ברשימת הפרויקטים, בוחרים את הפרויקט שרוצים למחוק ולוחצים על מחיקה
 .
. - כדי למחוק את הפרויקט, כותבים את מזהה הפרויקט בתיבת הדו-שיח ולוחצים על Shut down.