
1. परिचय
पिछले अपडेट की तारीख: 25-04-2022
आपको क्या बनाना है
इस कोडलैब में, Google Cloud पर अपने-आप स्केल होने वाला हाई परफ़ॉर्मेंस कंप्यूटिंग (एचपीसी) क्लस्टर डिप्लॉय किया जाएगा.Terraform डिप्लॉयमेंट, इस क्लस्टर को Spack के ज़रिए इंस्टॉल किए गए Gromacs के साथ बनाता है. क्लस्टर को Slurm जॉब शेड्यूलर की मदद से मैनेज किया जाएगा. क्लस्टर बन जाने के बाद, benchMEM, benchPEP या benchRIB बेंचमार्क चलाएं.
आपको क्या सीखने को मिलेगा
- Slurm जॉब शेड्यूलर के साथ काम करने वाले एचपीसी क्लस्टर को डिप्लॉय करने का तरीका
- Slurm बैच जॉब का इस्तेमाल करके, Google Cloud पर Gromacs की मदद से, जीपीयू की मदद से तेज़ किए गए मॉलिक्यूलर डाइनैमिक्स सिम्युलेशन को चलाने का तरीका
आपको इन चीज़ों की ज़रूरत होगी
2. सेटअप
इस कोडलैब को पूरा करने के लिए , आपको Compute Engine और Cloud Build API चालू करने होंगे. Compute Engine और Cloud Build API चालू करने के लिए, Cloud Shell खोलें और यहां दिए गए निर्देश चलाएं. नीचे दिए गए
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
चेतावनी: अगर आपको अपने क्लस्टर से कनेक्ट करने के लिए, तीसरे पक्ष के एसएसएच (जैसे, OpenSSH) का इस्तेमाल करना है, तो पक्का करें कि आपने ओएस लॉगिन का इस्तेमाल करके, अपनी Cloud Identity प्रोफ़ाइल में एसएसएच कुंजी अटैच की हो. अपनी Cloud Identity प्रोफ़ाइल में एसएसएच कुंजियां जोड़ने के बारे में ज़्यादा जानें.
3. [ज़रूरी नहीं] Gromacs के साथ GCP वीएम इमेज बनाना
इस कोडलैब के लिए, हमने आपके लिए पहले से बनी हुई इमेज उपलब्ध कराई है. ऐसा इसलिए, क्योंकि Gromacs और उससे जुड़ी सभी ज़रूरी चीज़ों को इंस्टॉल करने में दो घंटे लग सकते हैं. अगर आपको समय बचाने के लिए, पहले से बनी इस इमेज का इस्तेमाल करना है, तो अगले सेक्शन पर जाएं.
Google Cloud पर रिसर्च ऐप्लिकेशन चलाने के लिए, ऐप्लिकेशन को इंस्टॉल और डिप्लॉय करने के कई विकल्प उपलब्ध हैं. इस कोडलैब के इस सेक्शन में, आपको एक वर्चुअल मशीन इमेज बनानी होगी. यह slurm-gcp (CentOS7) वीएम इमेज पर आधारित होगी. बनाने की प्रोसेस के दौरान, कंपाइलर, Gromacs की सभी डिपेंडेंसी, और Gromacs इंस्टॉल हो जाएंगे.
RCC Apps repository पर मौजूद Gromacs Cloud Build pipeline में, Gromacs को इंस्टॉल करने के लिए ज़रूरी निर्देश शामिल होते हैं. इंस्टॉलेशन प्रोसेस में, Packer का इस्तेमाल करके एक वीएम डिप्लॉय किया जाता है. यह वीएम, Spack को इंस्टॉल करता है. इसके बाद, यह GCC@9.2.0 कंपाइलर और Gromacs@2021.2 को इंस्टॉल करता है. इसमें जीपीयू ऐक्सलरेशन की सुविधा चालू होती है.
- GCP पर Cloud Shell खोलें.
- FluidNumerics/rcc-apps रिपॉज़िटरी को क्लोन करें
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Google Cloud Build का इस्तेमाल करके इमेज बनाएं.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Google Cloud Build डैशबोर्ड पर जाकर, बिल्ड प्रोसेस की स्थिति देखी जा सकती है
बिल्ड करने की प्रोसेस में दो घंटे लग सकते हैं. इस प्रोसेस को तेज़ करने के लिए, अपनी बिल्ड कॉन्फ़िगरेशन फ़ाइल के स्कीमा में बदलाव करें, ताकि मशीन टाइप को बदला जा सके. इससे बिल्ड की परफ़ॉर्मेंस बेहतर होगी. इसके लिए, _MACHINE_TYPE बिल्ड वैरिएबल का इस्तेमाल किया जा सकता है. उदाहरण के लिए:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
बिल्ड पूरा होने के बाद, आपके Google Cloud प्रोजेक्ट में एक वीएम इमेज उपलब्ध होगी. इसका इस्तेमाल करके, क्लस्टर को डिप्लॉय किया जा सकता है.
4. Terraform की मदद से, ऑटो-स्केलिंग वाला एचपीसी क्लस्टर डिप्लॉय करना
इस सेक्शन में, आपको Slurm जॉब शेड्यूलर के साथ अपने-आप स्केल होने वाले एचपीसी क्लस्टर को डिप्लॉय करने के लिए, Terraform का इस्तेमाल करना होगा. इस क्लस्टर को ऐसे कंप्यूट नोड के साथ डिप्लॉय किया जाएगा जिनमें से हर एक में 8 वीसीपीयू और 1 Nvidia® Tesla V100 जीपीयू होगा.
- GCP पर Cloud Shell खोलें.
- FluidNumerics/rcc-apps रिपॉज़िटरी को क्लोन करें
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- gromacs terraform डायरेक्ट्री पर जाएं:
cd ~/rcc-apps/gromacs/tf/slurm
- टेराफ़ॉर्म प्लान बनाना और उसकी समीक्षा करना. अपने क्लस्टर, GCP प्रोजेक्ट, और उस ज़ोन का नाम तय करने के लिए, एनवायरमेंट वैरिएबल
GMX_NAME,GMX_PROJECT, औरGMX_ZONEसेट करें जहां आपको डिप्लॉय करना है. अगर आपको पक्का नहीं है, तो कृपया यहां दिया गया नोट पढ़ें
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- अगर आपने इस कोडलैब के पिछले सेक्शन में अपनी वीएम इमेज बनाई है, तो आपको GMX_IMAGE एनवायरमेंट वैरिएबल भी सेट करना होगा
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- मेक कमांड का इस्तेमाल करके प्लान बनाएं. इससे
terraform init && terraform planचलेगा.
make plan
- क्लस्टर डिप्लॉय करें. सेटअप प्रोसेस में सिर्फ़ कुछ मिनट लगते हैं, क्योंकि Gromacs और इसकी डिपेंडेंसी आपके क्लस्टर पर पहले से इंस्टॉल होती हैं.
make apply
- पिछले चरण में बनाए गए login नोड से SSH करें. आपको यह नोड पिछले चरण में दिखेगा. इसका नाम शायद gromacs-login0 होगा. इसके लिए, कंसोल मेन्यू आइटम Compute Engine -> वीएम इंस्टेंस में जाकर, वीएम इंस्टेंस की सूची के बगल में मौजूद एसएसएच बटन पर क्लिक करें.
विकल्प: gcloud कमांड की यह जोड़ी, लॉगिन नोड का नाम पता करेगी और इसमें एसएसएच करेगी:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- लॉगिन नोड से कनेक्ट होने के बाद, अपने क्लस्टर सेटअप की पुष्टि करने के लिए, देखें कि Gromacs इंस्टॉल है या नहीं
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- पुष्टि करें कि
/opt/share/gromacsमें यहां दिया गया कॉन्टेंट मौजूद है.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. benchRIB Benchmark को चलाएं
Gromacs, रिसर्च सॉफ़्टवेयर है. इसका इस्तेमाल मॉलिक्यूलर डाइनैमिक्स को सिम्युलेट करने और ऊर्जा को कम करने की सीमाओं के तहत मॉलिक्यूलर स्ट्रक्चर का हिसाब लगाने के लिए किया जाता है. इस कोडलैब के लिए वीएम इमेज में दिए गए बेंचमार्क, मॉलिक्यूलर डाइनैमिक्स पर फ़ोकस करते हैं. यह परमाणुओं के सिस्टम का विकास है.
मॉलिक्यूलर डाइनैमिक्स में, न्यूटन के गति के नियमों का इस्तेमाल करके, परमाणुओं की पोज़िशन, वेलोसिटी, और ऐक्सलरेशन को सिम्युलेट किया जाता है :
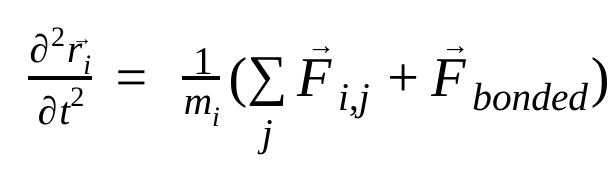
यहाँ, 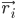 , ऐटम i की पोज़िशन है, t समय है,
, ऐटम i की पोज़िशन है, t समय है, 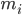 , ऐटम i का द्रव्यमान है, और
, ऐटम i का द्रव्यमान है, और 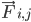 , ऐटम j की वजह से ऐटम i पर लगने वाला नॉन-बॉन्डेड फ़ोर्स है. साथ ही,
, ऐटम j की वजह से ऐटम i पर लगने वाला नॉन-बॉन्डेड फ़ोर्स है. साथ ही, 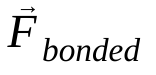 , बॉन्डेड इंटरैक्शन की वजह से लगने वाले फ़ोर्स हैं. तापमान, दबाव, परमाणु की पोज़िशन, और परमाणु की वेलोसिटी के आधार पर, फ़ोर्स का हिसाब लगाया जाता है. साथ ही, सिस्टम को संख्या के हिसाब से इंटिग्रेट किया जाता है, ताकि परमाणु की नई वेलोसिटी और पोज़िशन मिल सकें. इस प्रोसेस को दोहराया जाता है, ताकि किसी तय समय के लिए मॉलिक्यूलर डाइनैमिक्स को सिम्युलेट किया जा सके.
, बॉन्डेड इंटरैक्शन की वजह से लगने वाले फ़ोर्स हैं. तापमान, दबाव, परमाणु की पोज़िशन, और परमाणु की वेलोसिटी के आधार पर, फ़ोर्स का हिसाब लगाया जाता है. साथ ही, सिस्टम को संख्या के हिसाब से इंटिग्रेट किया जाता है, ताकि परमाणु की नई वेलोसिटी और पोज़िशन मिल सकें. इस प्रोसेस को दोहराया जाता है, ताकि किसी तय समय के लिए मॉलिक्यूलर डाइनैमिक्स को सिम्युलेट किया जा सके.
Gromacs इमेज (आपकी बनाई हुई या दी गई इमेज) में तीन बेंचमार्क होते हैं
- benchMEM
- benchRIB
- benchPEP
ये बेंचमार्क, डॉ. कुट्ज़नर के फ़्री Gromacs बेंचमार्क के सेट से लिए गए हैं. ये ट्रांज़िएंट मॉलिक्यूलर डाइनैमिक सिमुलेशन का स्टैंडर्ड सेट है. हर बेंचमार्क में, ऐटम की संख्या और सिम्युलेशन की अवधि अलग-अलग होती है. हर सिम्युलेशन के लिए ज़रूरी कॉन्फ़िगरेशन की जानकारी यहां दी गई है.
मेट्रिक / बेंचमार्क | benchMEM | benchRIB | benchPEP |
परमाणुओं की संख्या | 81,743 | 2,136,412 | 12,495,503 |
सिस्टम का साइज़ / नैनोमीटर | 10.8 x 10.2 x 9.6 | 31.2 x 31.2 x 31.2 | 50.0 x 50.0 x 50.0 |
टाइम स्टेप / fs | 2 | 4 | 2 |
कटऑफ़ रेडियस / एनएम | 1 | 1 | 1.2 |
PME ग्रिड स्पेसिंग / एनएम | 0.12 | 0.135 | 0.16 |
बेंचमार्क चलाने के लिए, आपको Slurm बैच जॉब सबमिट करना होगा. डिफ़ॉल्ट रूप से, दी गई बैच स्क्रिप्ट, benchRIB बेंचमार्क को चलाती है. जिन बेंचमार्क के लिए कॉन्फ़िगरेशन तय किए गए हैं उनके इनपुट डेक, Gromacs वीएम इमेज में /opt/share/gromacs के तहत शामिल किए गए हैं. इसके अलावा, Gromacs को चलाने के लिए bash स्क्रिप्ट का एक उदाहरण /opt/share में उपलब्ध है.
इस सेक्शन के लिए, आपको क्लस्टर के login नोड से SSH करना होगा
- sbatch कमांड का इस्तेमाल करके बैच जॉब सबमिट करना
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
इससे काम को पूरा करने के लिए, Slurm एक कंप्यूट नोड उपलब्ध कराएगा. sinfo कमांड चलाने पर, आपको दिखेगा कि कंप्यूट नोड alloc# स्थिति में है. इसका मतलब है कि कंप्यूट नोड को आपके काम के लिए असाइन कर दिया गया है, लेकिन उसे अभी चालू किया जा रहा है. जॉब शुरू होने के बाद, नोड को alloc स्थिति पर सेट कर दिया जाएगा.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
जॉब पूरा होने तक इंतज़ार करें. डिफ़ॉल्ट बेंचमार्क (benchRIB) में करीब 80 लाख एटम होते हैं. इसे 5,000 टाइमस्टेप (4 टाइमस्टेप/एफ़एस के साथ) चलाने के लिए कॉन्फ़िगर किया जाता है. इसे पूरा होने में करीब 6 मिनट लगते हैं. इस कमांड का इस्तेमाल करके, अपनी नौकरी की स्थिति देखी जा सकती है:
watch squeue
जब आपका काम कतार से हट जाए, तब ctrl-C दबाकर बाहर निकलें.
- टास्क पूरा होने के बाद, आपको
run/नाम की एक डायरेक्ट्री दिखेगी. इसमें सिम्युलेशन का आउटपुट (run/MEMमें) और आपकी मौजूदा डायरेक्ट्री मेंgromacs.outनाम की एक लॉग फ़ाइल होगी.run/MEMडायरेक्ट्री में दो फ़ाइलेंener.edrऔरmd.logहोती हैं.ener.edrफ़ाइल में, सिस्टम की ऊर्जा, तापमान, दबाव, बॉक्स का साइज़, घनत्व, और विरियल को पोर्टेबल बाइनरी फ़ॉर्मैट में सेव किया जाता है. एक्सटेंशन के सुझाव के मुताबिक,md.logफ़ाइल में Gromacs सिम्युलेशन के लॉग होते हैं. इसमें पार्टिकल-पार्टिकल और PME सॉल्वर से लॉगिंग की जानकारी के साथ-साथ, सिम्युलेशन की परफ़ॉर्मेंस के बारे में भी जानकारी शामिल होती है. gromacs.out फ़ाइल मेंgmx mdrunका स्टैंडर्ड आउटपुट होता है. यह नीचे दिखाए गए आउटपुट जैसा होना चाहिए.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. बधाई हो
इस कोडलैब में, आपने अपने-आप स्केल होने वाला, क्लाउड-नेटिव एचपीसी क्लस्टर बनाया. साथ ही, Google Cloud पर Gromacs की मदद से, जीपीयू की मदद से तेज़ी से काम करने वाले मॉलिक्यूलर डाइनैमिक्स सिमुलेशन को चलाया!
स्टोरेज खाली करना
इस कोडलैब में इस्तेमाल किए गए संसाधनों के लिए, अपने Google Cloud खाते से शुल्क न लिए जाने के लिए:
सुझाया गया: Terraform की मदद से एचपीसी क्लस्टर मिटाना
- Cloud Shell खोलें और
gromacs/tf/slurmउदाहरण डायरेक्ट्री पर जाएं
cd ~/rcc-apps/gromacs/tf/slurm
- सभी संसाधनों को मिटाने के लिए, make destroy कमांड चलाएं.
make destroy
या, प्रोजेक्ट मिटाएं (सबसे असरदार और नुकसान पहुंचाने वाला तरीका)
बिलिंग को बंद करने का सबसे आसान तरीका यह है कि आप उस प्रोजेक्ट को मिटा दें जिसे आपने कोडलैब के लिए बनाया था.
चेतावनी: किसी प्रोजेक्ट को मिटाने पर ये असर होते हैं:
- प्रोजेक्ट में मौजूद सभी आइटम मिट जाते हैं. अगर आपने इस कोडलैब के लिए किसी मौजूदा प्रोजेक्ट का इस्तेमाल किया है, तो उसे मिटाने पर, प्रोजेक्ट में किया गया आपका अन्य काम भी मिट जाएगा.
- कस्टम प्रोजेक्ट आईडी मिट जाते हैं. आपने इस प्रोजेक्ट को बनाते समय, कोई कस्टम प्रोजेक्ट आईडी बनाया होगा. आपको आने वाले समय में इसका इस्तेमाल करना है. प्रोजेक्ट आईडी का इस्तेमाल करने वाले यूआरएल को बनाए रखने के लिए, पूरे प्रोजेक्ट को मिटाने के बजाय, प्रोजेक्ट में मौजूद चुने गए संसाधनों को मिटाएं. जैसे, appspot.com यूआरएल.
अगर आपको कई कोडलैब और क्विकस्टार्ट आज़माने हैं, तो प्रोजेक्ट का दोबारा इस्तेमाल करने से, प्रोजेक्ट के कोटे की सीमाएं पार करने से बचा जा सकता है.
- Cloud Console में, संसाधन मैनेज करें पेज पर जाएं. संसाधन मैनेज करें पेज पर जाएं
- प्रोजेक्ट की सूची में, वह प्रोजेक्ट चुनें जिसे आपको मिटाना है. इसके बाद, मिटाएं
 पर क्लिक करें.
पर क्लिक करें. - डायलॉग बॉक्स में, प्रोजेक्ट आईडी टाइप करें. इसके बाद, प्रोजेक्ट मिटाने के लिए बंद करें पर क्लिक करें.