
1. Pengantar
Terakhir Diperbarui: 25-04-2022
Yang akan Anda buat
Dalam codelab ini, Anda akan men-deploy cluster Komputasi Performa Tinggi (HPC) penskalaan otomatis di Google Cloud.Deployment Terraform membuat cluster ini dengan Gromacs yang diinstal melalui Spack. Cluster akan dikelola dengan penjadwal tugas Slurm. Setelah cluster dibuat, Anda akan menjalankan benchmark benchMEM, benchPEP, atau benchRIB.
Yang akan Anda pelajari
- Cara men-deploy cluster HPC yang mendukung penjadwal tugas Slurm
- Cara menjalankan simulasi dinamika molekuler yang dipercepat GPU dengan Gromacs di Google Cloud menggunakan tugas batch Slurm
Yang Anda butuhkan
2. Penyiapan
Untuk menyelesaikan codelab ini , Anda perlu mengaktifkan Compute Engine API dan Cloud Build API. Untuk mengaktifkan Compute Engine API dan Cloud Build API, buka Cloud Shell dan jalankan perintah berikut. Jangan lupa untuk mengganti project-id Anda dengan
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Perhatian: Jika Anda berencana menggunakan SSH pihak ketiga (misalnya, OpenSSH) untuk terhubung ke cluster, pastikan Anda melampirkan kunci SSH ke profil identitas cloud menggunakan Login OS. Pelajari lebih lanjut cara menambahkan kunci SSH ke profil identitas cloud Anda.
3. [OPSIONAL] Buat Image VM GCP dengan Gromacs
Untuk codelab ini, kami telah menyediakan image yang telah dibuat sebelumnya untuk Anda, karena proses build dapat memakan waktu hingga 2 jam untuk menginstal Gromacs dan semua dependensinya. Jika ingin menggunakan image bawaan ini untuk menghemat waktu, Anda dapat langsung membuka bagian berikutnya.
Saat menjalankan aplikasi riset di Google Cloud, ada banyak opsi untuk menginstal dan men-deploy aplikasi Anda. Di bagian codelab ini, Anda akan membuat image virtual machine yang dibangun di atas image VM slurm-gcp (CentOS7). Selama proses pembuatan, compiler, semua dependensi Gromacs, dan Gromacs akan diinstal.
Pipeline Gromacs Cloud Build di repositori RCC Apps merangkum petunjuk yang diperlukan untuk menginstal Gromacs. Proses penginstalan menggunakan Packer untuk men-deploy VM yang menginstal Spack, yang selanjutnya menginstal compiler GCC@9.2.0 dan Gromacs@2021.2 dengan akselerasi GPU diaktifkan.
- Buka Cloud Shell di GCP.
- Buat clone repositori FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Bangun image menggunakan Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Anda dapat memeriksa status proses build di dasbor Google Cloud Build
Proses build dapat memerlukan waktu hingga dua jam. Untuk mempercepatnya,Anda dapat mempertimbangkan untuk mengubah skema file konfigurasi build guna mengubah jenis mesin untuk meningkatkan performa build. Hal ini dapat dilakukan menggunakan variabel build _MACHINE_TYPE. Contoh:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Setelah build selesai, Anda akan memiliki image VM yang tersedia di project Google Cloud yang dapat digunakan untuk men-deploy cluster.
4. Men-deploy cluster HPC penskalaan otomatis dengan Terraform
Di bagian ini, Anda akan menggunakan Terraform untuk men-deploy cluster HPC penskalaan otomatis, dengan penginstal penjadwal tugas Slurm. Cluster ini akan di-deploy dengan node komputasi yang masing-masing memiliki 8 vCPU dan 1 GPU Nvidia® Tesla V100.
- Buka Cloud Shell di GCP.
- Buat clone repositori FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Ubah ke direktori terraform gromacs:
cd ~/rcc-apps/gromacs/tf/slurm
- Buat dan tinjau rencana terraform. Tetapkan variabel lingkungan
GMX_NAME,GMX_PROJECT, danGMX_ZONEuntuk menentukan nama cluster, project GCP, dan zona tempat Anda ingin men-deploy. Tinjau catatan di bawah jika Anda tidak yakin
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Jika Anda membuat image VM sendiri di bagian sebelumnya dalam codelab ini, Anda juga perlu menyetel variabel lingkungan GMX_IMAGE
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Buat rencana dengan perintah make, yang akan menjalankan
terraform init && terraform plan.
make plan
- Deploy cluster. Proses penyiapan hanya memerlukan waktu beberapa menit karena Gromacs dan dependensinya sudah diinstal sebelumnya di cluster Anda.
make apply
- SSH ke node login yang dibuat pada langkah sebelumnya. Anda dapat melihat node ini di langkah sebelumnya (kemungkinan disebut gromacs-login0). Anda dapat melakukannya dengan mengklik tombol SSH di samping daftar Instance VM pada item menu konsol Compute Engine -> VM instance.
Opsi: Pasangan perintah gcloud ini akan mengetahui nama node login dan melakukan SSH ke node tersebut:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Setelah terhubung ke node login, untuk memverifikasi penyiapan cluster, periksa apakah Gromacs telah diinstal
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Verifikasi bahwa
/opt/share/gromacsmemiliki konten yang tercantum di bawah.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Menjalankan Benchmark benchRIB
Gromacs adalah software riset yang digunakan untuk menyimulasikan dinamika molekuler dan menghitung struktur molekuler dalam batasan minimisasi energi. Benchmark yang disediakan dalam image VM untuk codelab ini berfokus pada dinamika molekuler, evolusi sistem atom.
Dalam dinamika molekuler, posisi, kecepatan, dan percepatan atom disimulasikan menggunakan hukum gerak Newton :
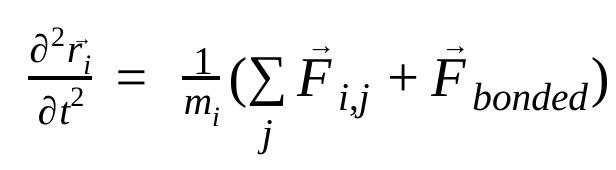
dengan 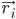 adalah posisi atom i, t adalah waktu,
adalah posisi atom i, t adalah waktu, 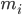 adalah massa atom i,
adalah massa atom i, 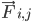 adalah gaya non-ikatan pada atom i akibat atom j, dan
adalah gaya non-ikatan pada atom i akibat atom j, dan 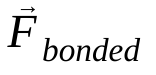 adalah gaya akibat interaksi ikatan. Dengan suhu, tekanan, posisi atom, dan kecepatan atom, gaya dihitung dan sistem diintegrasikan secara numerik untuk mendapatkan kecepatan dan posisi atom baru. Proses ini diulang untuk menyimulasikan dinamika molekuler selama jangka waktu tertentu.
adalah gaya akibat interaksi ikatan. Dengan suhu, tekanan, posisi atom, dan kecepatan atom, gaya dihitung dan sistem diintegrasikan secara numerik untuk mendapatkan kecepatan dan posisi atom baru. Proses ini diulang untuk menyimulasikan dinamika molekuler selama jangka waktu tertentu.
Gambar Gromacs (baik yang Anda buat maupun yang disediakan) dilengkapi dengan tiga tolok ukur
- benchMEM
- benchRIB
- benchPEP
Benchmark ini berasal dari kumpulan Benchmark Gromacs Gratis Dr. Kutzner dan merupakan kumpulan simulasi dinamika molekuler transien standar. Setiap tolok ukur bervariasi dalam jumlah atom dan durasi simulasi. Konfigurasi yang relevan untuk setiap simulasi diberikan dalam tabel di bawah.
Metrik / Benchmark | benchMEM | benchRIB | benchPEP |
Jumlah Atom | 81.743 | 2.136.412 | 12.495.503 |
Ukuran Sistem / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50,0 x 50,0 x 50,0 |
Time Step / fs | 2 | 4 | 2 |
Jari-jari Batas / nm | 1 | 1 | 1,2 |
Jarak petak PME / nm | 0,12 | 0,135 | 0,16 |
Untuk menjalankan tolok ukur, Anda akan mengirimkan tugas batch Slurm. Secara default, skrip batch yang disediakan menjalankan tolok ukur benchRIB. Set input yang menentukan konfigurasi untuk benchmark yang disediakan disertakan dalam image VM Gromacs di /opt/share/gromacs. Selain itu, contoh skrip bash untuk menjalankan Gromacs tersedia di /opt/share.
Untuk bagian ini, Anda harus menggunakan SSH ke node login cluster
- Mengirimkan tugas batch menggunakan perintah sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Tindakan ini akan mengantrekan tugas untuk dieksekusi dan Slurm akan menyediakan compute node untuk Anda. Saat menjalankan sinfo, Anda akan melihat bahwa node komputasi berada dalam status alloc#, yang berarti node komputasi dialokasikan untuk tugas Anda, tetapi sedang dalam proses penyediaan. Setelah tugas Anda berjalan, node akan ditetapkan ke status alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Tunggu hingga tugas selesai. Benchmark default (benchRIB) menyimulasikan sekitar 8 juta atom dan dikonfigurasi untuk menjalankan 5.000 langkah waktu (dengan 4 langkah waktu/fs) dan memerlukan waktu sekitar 6 menit untuk diselesaikan. Anda dapat memantau status tugas dengan perintah ini:
watch squeue
Setelah tugas Anda tidak ada lagi dalam antrean, Anda dapat keluar dengan ctrl-C.
- Setelah tugas selesai, Anda akan melihat direktori bernama
run/yang berisi output simulasi (dirun/MEM) dan file log di direktori saat ini bernamagromacs.out. Direktorirun/MEMberisi dua fileener.edrdanmd.log. Fileener.edrmenyimpan energi, suhu, tekanan, ukuran kotak, kepadatan, dan virials sistem dalam format biner portabel. Seperti yang disarankan oleh ekstensi, filemd.logberisi log untuk simulasi Gromacs dan menyertakan informasi tentang performa simulasi selain informasi logging dari pemecah masalah partikel-partikel dan PME. Isi gromacs.out yang berisi output standar darigmx mdrunakan terlihat mirip dengan yang ditampilkan di bawah.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Selamat
Dalam codelab ini, Anda telah membuat cluster HPC cloud-native dengan penskalaan otomatis dan menjalankan simulasi dinamika molekuler yang dipercepat GPU dengan Gromacs di Google Cloud.
Pembersihan
Agar tidak menimbulkan biaya pada akun Google Cloud Anda untuk resource yang digunakan dalam codelab ini:
DIREKOMENDASIKAN: Menghapus cluster HPC dengan Terraform
- Buka cloud shell Anda dan buka direktori contoh
gromacs/tf/slurm
cd ~/rcc-apps/gromacs/tf/slurm
- Jalankan make destroy untuk menghapus semua resource.
make destroy
ATAU, Hapus project (paling efektif dan merusak)
Cara termudah untuk menghilangkan penagihan adalah dengan menghapus project yang Anda buat untuk codelab.
Perhatian: Menghapus project akan mengakibatkan:
- Semua hal dalam project akan dihapus. Jika Anda menggunakan project yang sudah ada untuk codelab ini, saat Anda menghapusnya, pekerjaan lain dalam project tersebut juga akan dihapus.
- Project ID kustom akan hilang. Saat membuat project ini, Anda mungkin juga membuat project ID kustom yang masih ingin Anda gunakan pada masa mendatang. Agar tidak kehilangan URL yang menggunakan project ID tersebut, seperti URL appspot.com, hapus resource yang dipilih di dalam project, bukan menghapus seluruh project.
Jika Anda berencana mempelajari beberapa codelab dan panduan memulai, menggunakan kembali project dapat membantu Anda agar tidak melampaui batas kuota project.
- Di Cloud Console, buka halaman Manage resource. Buka halaman Kelola resource
- Dalam daftar project, pilih project yang ingin Anda hapus, lalu klik Hapus
 .
. - Pada dialog, ketik project ID, lalu klik Shut down untuk menghapus project.