
1. Introduzione
Ultimo aggiornamento: 25/04/2022
Cosa creerai
In questo codelab, eseguirai il deployment di un cluster di computing ad alte prestazioni (HPC) a scalabilità automatica su Google Cloud.Un deployment Terraform crea questo cluster con Gromacs installato tramite Spack. Il cluster verrà gestito con lo scheduler dei job Slurm. Una volta creato il cluster, esegui i benchmark benchMEM, benchPEP o benchRIB.
Cosa imparerai
- Come eseguire il deployment di un cluster HPC che supporta lo scheduler di job Slurm
- Come eseguire simulazioni di dinamica molecolare accelerate dalla GPU con Gromacs su Google Cloud utilizzando un job batch Slurm
Cosa ti serve
2. Configurazione
Per completare questo codelab , devi abilitare le API Compute Engine e Cloud Build. Per abilitare le API Compute Engine e Cloud Build, apri Cloud Shell ed esegui i seguenti comandi. Ricorda di sostituire project-id con
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Attenzione:se prevedi di utilizzare SSH di terze parti (ad es. OpenSSH) per connetterti al cluster, assicurati di collegare una chiave SSH al tuo profilo Cloud Identity utilizzando OS Login. Scopri di più sull'aggiunta di chiavi SSH al tuo profilo Cloud Identity.
3. [FACOLTATIVO] Crea un'immagine VM Google Cloud con Gromacs
Per questo codelab, ti abbiamo fornito un'immagine predefinita, poiché il processo di compilazione può richiedere fino a due ore per installare Gromacs e tutte le relative dipendenze. Se vuoi utilizzare questa immagine predefinita per risparmiare tempo, puoi passare alla sezione successiva.
Quando esegui applicazioni di ricerca su Google Cloud, hai molte opzioni per installare ed eseguire il deployment dell'applicazione. In questa sezione del codelab, creerai un'immagine di macchina virtuale basata sull'immagine VM slurm-gcp (CentOS7). Durante il processo di creazione, verranno installati il compilatore, tutte le dipendenze di Gromacs e Gromacs.
La pipeline di build cloud Gromacs nel repository RCC Apps contiene le istruzioni necessarie per installare Gromacs. La procedura di installazione utilizza Packer per eseguire il deployment di una VM che installa Spack, che a sua volta installa il compilatore GCC@9.2.0 e Gromacs@2021.2 con l'accelerazione GPU abilitata.
- Apri Cloud Shell su GCP.
- Clona il repository FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Crea l'immagine utilizzando Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Puoi controllare lo stato del processo di compilazione nella dashboard di Google Cloud Build.
La procedura di compilazione può richiedere fino a due ore. Per accelerare questo processo,puoi modificare lo schema del file di configurazione di compilazione per cambiare il tipo di macchina e migliorare il rendimento della compilazione. Questa operazione può essere eseguita utilizzando la variabile di build _MACHINE_TYPE. Ad esempio:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Al termine della build, nel tuo progetto Google Cloud sarà disponibile un'immagine VM che potrai utilizzare per eseguire il deployment del cluster.
4. Deployment di un cluster HPC a scalabilità automatica con Terraform
In questa sezione utilizzerai Terraform per eseguire il deployment di un cluster HPC a scalabilità automatica con lo scheduler dei job Slurm installato. Questo cluster verrà implementato con nodi di calcolo che dispongono ciascuno di 8 vCPU e 1 GPU Nvidia® Tesla V100.
- Apri Cloud Shell su GCP.
- Clona il repository FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Passa alla directory Terraform di GROMACS:
cd ~/rcc-apps/gromacs/tf/slurm
- Crea e rivedi un piano Terraform. Imposta le variabili di ambiente
GMX_NAME,GMX_PROJECTeGMX_ZONEper specificare il nome del cluster, il progetto GCP e la zona in cui vuoi eseguire il deployment. Se non hai la certezza, consulta la nota riportata di seguito.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Se hai creato la tua immagine VM nella sezione precedente di questo codelab, devi anche impostare la variabile di ambiente GMX_IMAGE
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Crea il piano con il comando make, che eseguirà
terraform init && terraform plan.
make plan
- Esegui il deployment del cluster. La procedura di configurazione richiede solo pochi minuti, poiché Gromacs e le relative dipendenze sono preinstallati sul cluster.
make apply
- Connettiti tramite SSH al nodo login creato nel passaggio precedente. Puoi vedere questo nodo nel passaggio precedente (probabilmente chiamato gromacs-login0). Puoi farlo facendo clic sul pulsante SSH accanto all'elenco delle istanze VM nella voce di menu della console Compute Engine -> Istanza VM.
Opzione:questa coppia di comandi gcloud determina il nome del nodo di accesso e vi accede tramite SSH:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Una volta connesso al nodo di accesso, per verificare la configurazione del cluster, controlla che Gromacs sia installato
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Verifica che
/opt/share/gromacscontenga i contenuti elencati di seguito.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Eseguire il benchmark benchRIB
Gromacs è un software di ricerca utilizzato per simulare la dinamica molecolare e calcolare le strutture molecolari in base a vincoli di minimizzazione dell'energia. I benchmark forniti nell'immagine VM per questo codelab si concentrano sulla dinamica molecolare, l'evoluzione dei sistemi di atomi.
Nella dinamica molecolare, le posizioni, le velocità e le accelerazioni degli atomi vengono simulate utilizzando le leggi del moto di Newton :
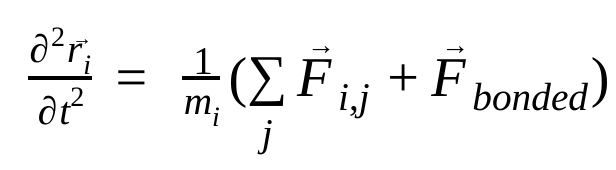
dove 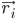 è la posizione dell'atomo i, t è il tempo,
è la posizione dell'atomo i, t è il tempo, 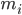 è la massa dell'atomo i e
è la massa dell'atomo i e 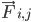 è la forza non legata sull'atomo i dovuta all'atomo j e
è la forza non legata sull'atomo i dovuta all'atomo j e 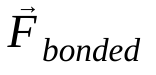 sono le forze dovute alle interazioni legate. Date la temperatura, la pressione, le posizioni degli atomi e le loro velocità, le forze vengono calcolate e il sistema viene integrato numericamente per ottenere nuove velocità e posizioni degli atomi. Questa procedura viene ripetuta per simulare la dinamica molecolare per un determinato periodo di tempo.
sono le forze dovute alle interazioni legate. Date la temperatura, la pressione, le posizioni degli atomi e le loro velocità, le forze vengono calcolate e il sistema viene integrato numericamente per ottenere nuove velocità e posizioni degli atomi. Questa procedura viene ripetuta per simulare la dinamica molecolare per un determinato periodo di tempo.
L'immagine Gromacs (quella che hai creato o quella fornita) include tre benchmark
- benchMEM
- benchRIB
- benchPEP
Questi benchmark provengono dal set di benchmark Gromacs senza costi del Dr. Kutzner e sono un insieme standard di simulazioni di dinamica molecolare transitoria. Ogni benchmark varia in base al numero di atomi e alla durata della simulazione. Le configurazioni pertinenti per ogni simulazione sono riportate nella tabella seguente.
Metrica / Benchmark | benchMEM | benchRIB | benchPEP |
Numero di atomi | 81.743 | 2.136.412 | 12.495.503 |
Dimensioni sistema / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50,0 x 50,0 x 50,0 |
Time Step / fs | 2 | 4 | 2 |
Raggi di taglio / nm | 1 | 1 | 1,2 |
Spaziatura griglia PME / nm | 0,12 | 0,135 | 0,16 |
Per eseguire il benchmark, invierai un job batch Slurm. Per impostazione predefinita, lo script batch fornito esegue il benchmark benchRIB. I mazzi di input che definiscono le configurazioni per i benchmark forniti sono inclusi nell'immagine VM Gromacs in /opt/share/gromacs. Inoltre, in /opt/share è disponibile uno script bash di esempio per l'esecuzione di Gromacs.
Per questa sezione, devi utilizzare SSH per connetterti al nodo login del cluster.
- Invia un job batch utilizzando il comando sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Il job verrà messo in coda per l'esecuzione e Slurm eseguirà il provisioning di un nodo di calcolo per te. Quando esegui sinfo, vedrai che un nodo di calcolo è nello stato alloc#, il che significa che il nodo di calcolo è allocato per il tuo job, ma è in fase di provisioning. Una volta eseguito il job, il nodo verrà impostato sullo stato alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Attendi il completamento del job. Il benchmark predefinito (benchRIB) simula circa 8 milioni di atomi ed è configurato per eseguire 5000 passaggi di tempo (con 4 passaggi di tempo/fs) e richiede circa 6 minuti per essere completato. Puoi monitorare lo stato del job con questo comando:
watch squeue
Quando il job non è più nella coda, puoi uscire con Ctrl+C.
- Al termine del job, dovresti visualizzare una directory denominata
run/che contiene l'output della simulazione (inrun/MEM) e un file di log nella directory corrente denominatogromacs.out. La directoryrun/MEMcontiene due file:ener.edremd.log. Il fileener.edrmemorizza le energie, la temperatura, la pressione, le dimensioni della scatola, la densità e i viriali del sistema in un formato binario portatile. Come suggerisce l'estensione, il filemd.logcontiene i log della simulazione Gromacs e include informazioni sulle prestazioni della simulazione, oltre alle informazioni di logging dei risolutori particella-particella e PME. I contenuti di gromacs.out contengono l'output standard digmx mdrune dovrebbero essere simili a quelli mostrati di seguito.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Complimenti
In questo codelab hai creato un cluster HPC cloud-native con scalabilità automatica ed eseguito una simulazione di dinamica molecolare con accelerazione GPU con Gromacs su Google Cloud.
Pulizia
Per evitare che al tuo account Google Cloud vengano addebitati costi relativi alle risorse utilizzate in questo codelab:
CONSIGLIATO: elimina il cluster HPC con Terraform
- Apri Cloud Shell e vai alla directory di esempio
gromacs/tf/slurm
cd ~/rcc-apps/gromacs/tf/slurm
- Esegui make destroy per eliminare tutte le risorse.
make destroy
OPPURE, elimina il progetto (il metodo più efficace e distruttivo)
Il modo più semplice per eliminare la fatturazione è eliminare il progetto creato per il codelab.
Attenzione: l'eliminazione di un progetto ha i seguenti effetti:
- L'intero contenuto del progetto viene eliminato. Se hai utilizzato un progetto esistente per questo codelab, quando lo eliminerai, eliminerai anche tutto il lavoro che hai svolto nel progetto.
- Gli ID progetto personalizzati non sono più disponibili. Quando hai creato questo progetto, potresti aver creato un ID progetto personalizzato che vuoi utilizzare in futuro. Per conservare gli URL che utilizzano l'ID progetto, ad esempio un URL appspot.com, elimina le risorse selezionate all'interno del progetto anziché eliminare l'intero progetto.
Se intendi esplorare più codelab e guide rapide, puoi riutilizzare i progetti ed evitare così di superare i limiti di quota.
- In Cloud Console, vai alla pagina Gestisci risorse. Vai alla pagina Gestisci risorse
- Nell'elenco dei progetti, seleziona il progetto che vuoi eliminare, quindi fai clic su Elimina
 .
. - Nella finestra di dialogo, digita l'ID progetto, quindi fai clic su Chiudi per eliminare il progetto.