
1. はじめに
最終更新日: 2022 年 4 月 25 日
作業内容
この Codelab では、Google Cloud に自動スケーリング ハイ パフォーマンス コンピューティング(HPC)クラスタをデプロイします。Terraform デプロイにより、Spack を介して Gromacs がインストールされたクラスタが作成されます。クラスタは Slurm ジョブ スケジューラで管理されます。クラスタが作成されたら、benchMEM、benchPEP、benchRIB のベンチマークを実行します。
学習内容
- Slurm ジョブ スケジューラをサポートする HPC クラスタをデプロイする方法
- Slurm バッチジョブを使用して Google Cloud で Gromacs を使用して GPU アクセラレーションによる分子動力学シミュレーションを実行する方法
必要なもの
2. セットアップ
この Codelab を完了するには、Compute Engine API と Cloud Build API を有効にする必要があります。Compute Engine API と Cloud Build API を有効にするには、Cloud Shell を開いて次のコマンドを実行します。下の
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
注意: サードパーティの SSH(OpenSSH など)を使用してクラスタに接続する場合は、OS Login を使用してクラウド ID プロファイルに SSH 認証鍵を関連付けてください。Cloud Identity プロファイルに SSH 認証鍵を追加する方法の詳細を確認する。
3. [省略可] Gromacs を使用して GCP VM イメージを作成する
この Codelab では、ビルド プロセスで Gromacs とそのすべての依存関係のインストールに最大 2 時間かかるため、ビルド済みのイメージを用意しています。この事前構築済みイメージを使用して時間を節約する場合は、次のセクションに進んでください。
Google Cloud で研究アプリケーションを実行する場合、アプリケーションのインストールとデプロイには多くのオプションがあります。このセクションでは、slurm-gcp(CentOS7)VM イメージをベースに構築する仮想マシン イメージを作成します。作成プロセスでは、コンパイラ、Gromacs のすべての依存関係、Gromacs がインストールされます。
RCC Apps リポジトリの Gromacs Cloud Build パイプラインには、Gromacs のインストールに必要な手順がカプセル化されています。 インストール プロセスでは、Packer を使用して Spack をインストールする VM をデプロイします。これにより、GPU アクセラレーションが有効になった GCC@9.2.0 コンパイラと Gromacs@2021.2 がインストールされます。
- GCP で Cloud Shell を開きます。
- FluidNumerics/rcc-apps リポジトリのクローンを作成する
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Google Cloud Build を使用してイメージをビルドします。
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
ビルドプロセスのステータスは、Google Cloud Build ダッシュボードで確認できます。
ビルドプロセスには最大 2 時間ほどかかることがあります。これを高速化するには、ビルド パフォーマンスを改善するためにマシンタイプを変更するよう、ビルド構成ファイル スキーマの変更を検討してください。これは、_MACHINE_TYPE ビルド変数を使用して行うことができます。次に例を示します。
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
ビルドが完了すると、クラスタのデプロイに使用できる VM イメージが Google Cloud プロジェクトで使用可能になります。
4. Terraform を使用して自動スケーリング HPC クラスタをデプロイする
このセクションでは、Terraform を使用して、Slurm ジョブ スケジューラがインストールされた自動スケーリング HPC クラスタをデプロイします。このクラスタは、それぞれ 8 個の vCPU と 1 個の Nvidia® Tesla V100 GPU を持つコンピューティング ノードでデプロイされます。
- GCP で Cloud Shell を開きます。
- FluidNumerics/rcc-apps リポジトリのクローンを作成する
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- gromacs terraform ディレクトリに移動します。
cd ~/rcc-apps/gromacs/tf/slurm
- Terraform プランを作成して確認します。環境変数
GMX_NAME、GMX_PROJECT、GMX_ZONEを設定して、クラスタの名前、GCP プロジェクト、デプロイ先のゾーンを指定します。ご不明な場合は、以下の注をご覧ください。
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- この Codelab の前のセクションで独自の VM イメージを作成した場合は、GMX_IMAGE 環境変数も設定する必要があります。
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- make コマンドを使用してプランを作成します。これにより、
terraform init && terraform planが実行されます。
make plan
- クラスタをデプロイします。Gromacs とその依存関係はクラスタにプリインストールされているため、設定プロセスには数分しかかかりません。
make apply
- 前の手順で作成したログインノードに SSH で接続します。このノードは前の手順で確認できます(おそらく gromacs-login0 という名前です)。これを行うには、コンソール メニュー項目の [Compute Engine] -> [VM インスタンス] の VM インスタンスのリストの横にある [SSH] ボタンをクリックします。
オプション: 次の gcloud コマンドのペアは、ログインノード名を特定して SSH で接続します。
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- ログインノードに接続したら、クラスタの設定を確認するために、Gromacs がインストールされていることを確認します。
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
/opt/share/gromacsに以下の内容が含まれていることを確認します。
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. benchRIB ベンチマークを実行する
Gromacs は、分子動力学のシミュレーションと、エネルギー最小化制約下での分子構造の計算に使用される研究ソフトウェアです。この Codelab の VM イメージで提供されるベンチマークは、分子動力学、つまり原子系の進化に重点を置いています。
分子動力学では、ニュートンの運動法則を使用して原子の位置、速度、加速度をシミュレートします。
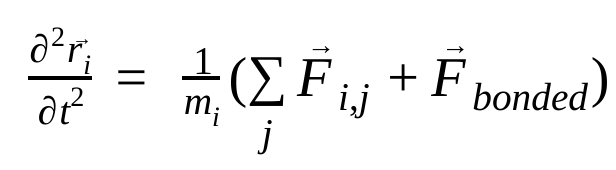
ここで、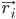 は原子 i の位置、t は時間、
は原子 i の位置、t は時間、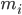 は原子 i の質量、
は原子 i の質量、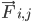 は原子 j による原子 i の非結合力、
は原子 j による原子 i の非結合力、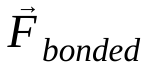 は結合相互作用による力です。温度、圧力、原子の位置、原子の速度が与えられると、力が計算され、システムが数値的に積分されて、新しい原子の速度と位置が求められます。このプロセスを繰り返して、特定の期間の分子動力学をシミュレートします。
は結合相互作用による力です。温度、圧力、原子の位置、原子の速度が与えられると、力が計算され、システムが数値的に積分されて、新しい原子の速度と位置が求められます。このプロセスを繰り返して、特定の期間の分子動力学をシミュレートします。
Gromacs イメージ(作成したイメージまたは提供されたイメージ)には 3 つのベンチマークが付属しています。
- benchMEM
- benchRIB
- benchPEP
これらのベンチマークは、Dr. Kutzner の Free Gromacs Benchmarks セット のもので、標準的な一過性の分子動力学シミュレーションのセットです。各ベンチマークは、原子の数とシミュレーションの期間が異なります。各シミュレーションの関連する構成を次の表に示します。
指標 / ベンチマーク | benchMEM | benchRIB | benchPEP |
Number of Atoms | 81,743 | 2,136,412 | 12,495,503 |
System Size / nm | 10.8 x 10.2 x 9.6 | 31.2 x 31.2 x 31.2 | 50.0 x 50.0 x 50.0 |
タイムステップ / fs | 2 | 4 | 2 |
カットオフ半径 / nm | 1 | 1 | 1.2 |
PME グリッド間隔 / nm | 0.12 | 0.135 | 0.16 |
ベンチマークを実行するには、Slurm バッチジョブを送信します。デフォルトでは、提供されたバッチ スクリプトは benchRIB ベンチマークを実行します。提供されたベンチマークの構成を定義する入力デッキは、Gromacs VM イメージの /opt/share/gromacs に含まれています。また、Gromacs を実行するための bash スクリプトの例は /opt/share にあります。
このセクションでは、クラスタのログインノードに SSH で接続する必要があります。
- sbatch コマンドを使用してバッチジョブを送信する
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
これにより、ジョブが実行待ち状態になり、Slurm がコンピューティング ノードをプロビジョニングします。sinfo を実行すると、コンピューティング ノードが alloc# 状態になっていることがわかります。これは、コンピューティング ノードがジョブに割り当てられているものの、プロビジョニング中であることを意味します。ジョブが実行されると、ノードは alloc 状態に設定されます。
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
ジョブが完了するまで、デフォルトのベンチマーク(benchRIB)は約 800 万個の原子をシミュレートし、5,000 タイムステップ(4 タイムステップ/fs)ステップを実行するように構成されており、完了までに約 6 分かかります。次のコマンドを使用して、ジョブのステータスをモニタリングできます。
watch squeue
ジョブがキューから消えたら、Ctrl+C で終了できます。
- ジョブが完了すると、現在のディレクトリに、シミュレーション出力(
run/MEMの下)を含むrun/というディレクトリと、gromacs.outというログファイルが表示されます。run/MEMディレクトリには、ener.edrとmd.logの 2 つのファイルが含まれています。ener.edrファイルには、システムのエネルギー、温度、圧力、ボックスサイズ、密度、ビリアルがポータブルなバイナリ形式で保存されます。拡張子が示すように、md.logファイルには Gromacs シミュレーションのログが含まれており、粒子間ソルバーと PME ソルバーからのロギング情報に加えて、シミュレーションのパフォーマンスに関する情報も含まれています。gromacs.out の内容は、gmx mdrunからの標準出力で、次のようになります。
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. 完了
この Codelab では、自動スケーリングのクラウド ネイティブ HPC クラスタを作成し、Google Cloud で Gromacs を使用して GPU アクセラレータによる分子動力学シミュレーションを実行しました。
クリーンアップ
この Codelab で使用したリソースについて、Google Cloud アカウントに課金されないようにする手順は次のとおりです。
推奨: Terraform を使用して HPC クラスタを削除する
- Cloud Shell を開き、
gromacs/tf/slurmのサンプル ディレクトリに移動します。
cd ~/rcc-apps/gromacs/tf/slurm
- make destroy を実行して、すべてのリソースを削除します。
make destroy
または、プロジェクトを削除する(最も効果的で破壊的)
課金を停止する最も簡単な方法は、この Codelab 用に作成したプロジェクトを削除することです。
注意: プロジェクトを削除すると、次のような影響があります。
- プロジェクト内のすべてのものが削除されます。この Codelab で既存のプロジェクトを使用した場合、それを削除すると、そのプロジェクトで行った他の作業もすべて削除されます。
- カスタム プロジェクト ID が失われます。このプロジェクトを作成したときに、将来使用するカスタム プロジェクト ID を作成した可能性があります。appspot.com URL など、プロジェクト ID を使用する URL を保持するには、プロジェクト全体を削除するのではなく、プロジェクト内の選択したリソースを削除します。
複数の Codelab とクイックスタートを実施する予定がある場合は、プロジェクトを再利用すると、プロジェクトの割り当て上限を超えないようにできます。
- Cloud コンソールで [リソースの管理] ページに移動します。[リソースの管理] ページに移動
- プロジェクト リストで、削除するプロジェクトを選択し、[削除 ]
 をクリックします。
をクリックします。 - ダイアログでプロジェクト ID を入力し、[シャットダウン] をクリックしてプロジェクトを削除します。