
1. 소개
최종 업데이트: 2022-04-25
빌드할 항목
이 Codelab에서는 Google Cloud에 자동 확장 고성능 컴퓨팅 (HPC) 클러스터를 배포합니다.Terraform 배포는 Spack을 통해 Gromacs가 설치된 이 클러스터를 만듭니다. 클러스터는 Slurm 작업 스케줄러로 관리됩니다. 클러스터가 생성되면 benchMEM, benchPEP 또는 benchRIB 벤치마크를 실행합니다.
학습할 내용
- Slurm 작업 스케줄러를 지원하는 HPC 클러스터를 배포하는 방법
- Slurm 일괄 작업을 사용하여 Google Cloud에서 Gromacs로 GPU 가속 분자 역학 시뮬레이션을 실행하는 방법
필요한 항목
2. 설정
이 Codelab을 완료하려면 Compute Engine 및 Cloud Build API를 사용 설정해야 합니다. Compute Engine 및 Cloud Build API를 사용 설정하려면 Cloud Shell을 열고 다음 명령어를 실행합니다. 아래의
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
주의: 서드 파티 SSH (예: OpenSSH)를 사용하여 클러스터에 연결하려면 OS 로그인을 사용하여 클라우드 ID 프로필에 SSH 키를 연결해야 합니다. 클라우드 ID 프로필에 SSH 키를 추가하는 방법 자세히 알아보기
3. [선택사항] Gromacs로 GCP VM 이미지 만들기
이 Codelab에서는 빌드 프로세스에서 Gromacs 및 모든 종속 항목을 설치하는 데 최대 2시간이 걸릴 수 있으므로 미리 빌드된 이미지를 제공합니다. 이 미리 빌드된 이미지를 사용하여 시간을 절약하려면 다음 섹션으로 건너뛰세요.
Google Cloud에서 연구 애플리케이션을 실행할 때 애플리케이션을 설치하고 배포하는 다양한 옵션이 있습니다. 이 Codelab 섹션에서는 slurm-gcp (CentOS7) VM 이미지를 기반으로 하는 가상 머신 이미지를 만듭니다. 생성 프로세스 중에 컴파일러, 모든 Gromacs 종속 항목, Gromacs가 설치됩니다.
RCC Apps 저장소의 Gromacs Cloud Build 파이프라인은 Gromacs 설치에 필요한 안내를 캡슐화합니다. 설치 프로세스는 Packer를 사용하여 Spack을 설치하는 VM을 배포합니다. Spack은 GPU 가속이 사용 설정된 GCC@9.2.0 컴파일러와 Gromacs@2021.2를 설치합니다.
- GCP에서 Cloud Shell을 엽니다.
- FluidNumerics/rcc-apps 저장소를 클론합니다.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Google Cloud Build를 사용하여 이미지를 빌드합니다.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Google Cloud Build 대시보드에서 빌드 프로세스의 상태를 확인할 수 있습니다.
빌드 프로세스는 최대 2시간이 걸릴 수 있습니다. 이를 가속화하려면 빌드 성능을 개선하기 위해 머신 유형을 변경하도록 빌드 구성 파일 스키마를 수정하는 것이 좋습니다. 이는 _MACHINE_TYPE 빌드 변수를 사용하여 수행할 수 있습니다. 예를 들면 다음과 같습니다.
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
빌드가 완료되면 클러스터를 배포하는 데 사용할 수 있는 VM 이미지가 Google Cloud 프로젝트에 제공됩니다.
4. Terraform을 사용하여 자동 확장 HPC 클러스터 배포
이 섹션에서는 Terraform을 사용하여 Slurm 작업 스케줄러가 설치된 자동 확장 HPC 클러스터를 배포합니다. 이 클러스터는 각각 vCPU 8개와 Nvidia® Tesla V100 GPU 1개가 있는 컴퓨팅 노드로 배포됩니다.
- GCP에서 Cloud Shell을 엽니다.
- FluidNumerics/rcc-apps 저장소를 클론합니다.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- gromacs terraform 디렉터리로 변경합니다.
cd ~/rcc-apps/gromacs/tf/slurm
- Terraform 계획을 만들고 검토합니다.
GMX_NAME,GMX_PROJECT,GMX_ZONE환경 변수를 설정하여 클러스터 이름, GCP 프로젝트, 배포할 영역을 지정합니다. 잘 모르는 경우 아래 참고사항을 검토하세요.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- 이 Codelab의 이전 섹션에서 자체 VM 이미지를 만든 경우 GMX_IMAGE 환경 변수도 설정해야 합니다.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
terraform init && terraform plan을 실행하는 make 명령어로 계획을 만듭니다.
make plan
- 클러스터를 배포합니다. Gromacs와 종속 항목이 클러스터에 사전 설치되어 있으므로 설정 프로세스는 몇 분밖에 걸리지 않습니다.
make apply
- 이전 단계에서 만든 login 노드로 SSH합니다. 이전 단계에서 이 노드를 확인할 수 있습니다 (아마도 gromacs-login0). 콘솔 메뉴 항목 Compute Engine -> VM 인스턴스 의 VM 인스턴스 목록 옆에 있는 SSH 버튼을 클릭하여 이 작업을 실행할 수 있습니다.
옵션: 이 gcloud 명령어 쌍은 로그인 노드 이름을 파악하고 SSH합니다.
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- 로그인 노드에 연결되면 클러스터 설정을 확인하기 위해 Gromacs가 설치되어 있는지 확인합니다.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
/opt/share/gromacs에 아래 나열된 콘텐츠가 있는지 확인합니다.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. benchRIB 벤치마크 실행
Gromacs는 분자 역학을 시뮬레이션하고 에너지 최소화 제약 조건에서 분자 구조를 계산하는 데 사용되는 연구 소프트웨어입니다. 이 Codelab의 VM 이미지에 제공된 벤치마크는 분자 역학, 즉 원자 시스템의 진화에 중점을 둡니다.
분자 역학에서 원자의 위치, 속도, 가속도는 뉴턴의 운동 법칙을 사용하여 시뮬레이션됩니다.

여기서 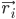 는 원자 i의 위치이고, t는 시간이며,
는 원자 i의 위치이고, t는 시간이며, 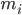 는 원자 i의 질량이고
는 원자 i의 질량이고 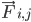 는 원자 j로 인한 원자 i의 비결합력이며
는 원자 j로 인한 원자 i의 비결합력이며 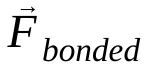 는 결합된 상호작용으로 인한 힘입니다. 온도, 압력, 원자 위치, 원자 속도가 주어지면 힘이 계산되고 시스템이 수치적으로 통합되어 새로운 원자 속도와 위치를 얻습니다. 이 프로세스는 지정된 기간 동안 분자 역학을 시뮬레이션하기 위해 반복됩니다.
는 결합된 상호작용으로 인한 힘입니다. 온도, 압력, 원자 위치, 원자 속도가 주어지면 힘이 계산되고 시스템이 수치적으로 통합되어 새로운 원자 속도와 위치를 얻습니다. 이 프로세스는 지정된 기간 동안 분자 역학을 시뮬레이션하기 위해 반복됩니다.
Gromacs 이미지 (직접 만든 이미지 또는 제공된 이미지)에는 세 가지 벤치마크가 포함되어 있습니다.
- benchMEM
- benchRIB
- benchPEP
이러한 벤치마크는 Kutzner 박사의 무료 Gromacs 벤치마크 세트 에서 가져온 것으로, 일시적인 분자 역학 시뮬레이션의 표준 세트입니다. 각 벤치마크는 원자 수와 시뮬레이션 기간이 다릅니다. 각 시뮬레이션과 관련된 구성은 아래 표에 나와 있습니다.
측정항목 / 벤치마크 | benchMEM | benchRIB | benchPEP |
원자 수 | 81,743 | 2,136,412 | 12,495,503 |
시스템 크기 / nm | 10.8 x 10.2 x 9.6 | 31.2 x 31.2 x 31.2 | 50.0 x 50.0 x 50.0 |
시간 단계 / fs | 2 | 4 | 2 |
차단 반지름 / nm | 1 | 1 | 1.2 |
PME 그리드 간격 / nm | 0.12 | 0.135 | 0.16 |
벤치마크를 실행하려면 Slurm 일괄 작업 을 제출합니다. 기본적으로 제공된 일괄 스크립트는 benchRIB 벤치마크를 실행합니다. 제공된 벤치마크의 구성을 정의하는 입력 데크는 /opt/share/gromacs 아래의 Gromacs VM 이미지에 포함되어 있습니다. 또한 Gromacs를 실행하는 Bash 스크립트 예는 /opt/share에서 확인할 수 있습니다.
이 섹션에서는 클러스터의 login 노드로 SSH해야 합니다.
- sbatch 명령어를 사용하여 일괄 작업을 제출합니다.
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
이렇게 하면 작업이 실행 대기열에 추가되고 Slurm이 컴퓨팅 노드를 프로비저닝합니다. sinfo를 실행하면 컴퓨팅 노드가 alloc# 상태인 것을 확인할 수 있습니다. 즉, 컴퓨팅 노드가 작업에 할당되었지만 프로비저닝 중입니다. 작업이 실행되면 노드가 alloc 상태로 설정됩니다.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
작업이 완료될 때까지 기다립니다. 기본 벤치마크 (benchRIB)는 약 8백만 개의 원자를 시뮬레이션하며 5,000개의 시간 단계 (시간 단계 4개/fs)를 실행하고 완료하는 데 약 6분이 걸리도록 구성됩니다. 이 명령어를 사용하여 작업 상태를 모니터링할 수 있습니다.
watch squeue
작업이 대기열에서 사라지면 Ctrl+C를 눌러 종료할 수 있습니다.
- 작업이 완료되면 시뮬레이션 출력이 포함된
run/디렉터리 (run/MEM아래)와 현재 디렉터리에gromacs.out이라는 로그 파일이 표시됩니다.run/MEM디렉터리에는ener.edr및md.log라는 두 개의 파일이 포함되어 있습니다.ener.edr파일은 시스템의 에너지, 온도, 압력, 상자 크기, 밀도, 비리알을 휴대용 바이너리 형식으로 저장합니다. 확장 프로그램에서 알 수 있듯이md.log파일에는 Gromacs 시뮬레이션의 로그가 포함되어 있으며 입자-입자 및 PME 솔버의 로깅 정보 외에도 시뮬레이션 성능에 관한 정보가 포함되어 있습니다. gromacs.out의 콘텐츠에는gmx mdrun의 표준 출력이 포함되어 있으며 아래와 유사하게 표시됩니다.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. 축하합니다
이 Codelab에서는 자동 확장 클라우드 네이티브 HPC 클러스터를 만들고 Google Cloud에서 Gromacs로 GPU 가속 분자 역학 시뮬레이션을 실행했습니다.
삭제
이 Codelab에서 사용한 리소스 비용이 Google Cloud 계정에 청구되지 않도록 하려면 다음 안내를 따르세요.
권장사항: Terraform으로 HPC 클러스터 삭제
- Cloud Shell을 열고
gromacs/tf/slurm예시 디렉터리로 이동합니다.
cd ~/rcc-apps/gromacs/tf/slurm
- make destroy를 실행하여 모든 리소스를 삭제합니다.
make destroy
또는 프로젝트 삭제 (가장 효과적이고 파괴적임)
비용이 청구되지 않도록 하는 가장 쉬운 방법은 Codelab에서 만든 프로젝트를 삭제하는 것입니다.
주의: 프로젝트를 삭제하면 다음과 같은 효과가 발생합니다.
- 프로젝트의 모든 항목이 삭제됩니다. 이 Codelab에서 기존 프로젝트를 사용한 경우 프로젝트를 삭제하면 프로젝트에서 수행한 다른 작업도 삭제됩니다.
- 커스텀 프로젝트 ID가 손실됩니다. 이 프로젝트를 만들 때 앞으로 사용할 커스텀 프로젝트 ID를 만들었을 수 있습니다. appspot.com URL과 같이 프로젝트 ID를 사용하는 URL을 보존하려면 전체 프로젝트를 삭제하는 대신 프로젝트 내의 선택된 리소스만 삭제하세요.
여러 Codelab과 빠른 시작을 살펴보려는 경우 프로젝트를 재사용하면 프로젝트 할당량 한도 초과를 방지할 수 있습니다.
- Cloud 콘솔에서 리소스 관리 페이지로 이동합니다. 리소스 관리 페이지로 이동
- 프로젝트 목록에서 삭제할 프로젝트를 선택하고 삭제
 를 클릭합니다.
를 클릭합니다. - 대화상자에서 프로젝트 ID를 입력한 후 종료 를 클릭하여 프로젝트를 삭제합니다.