
1. Введение
Последнее обновление: 25.04.2022
Что вы построите
В этом практическом задании вы развернете кластер высокопроизводительных вычислений (HPC) с автоматическим масштабированием в Google Cloud. Для создания этого кластера с установленным через Spack пакетом Gromacs используется Terraform. Управление кластером будет осуществляться с помощью планировщика заданий Slurm. После создания кластера вы запустите тесты benchMEM, benchPEP или benchRIB .
Что вы узнаете
- Как развернуть высокопроизводительный вычислительный кластер с поддержкой планировщика заданий Slurm.
- Как запустить ускоренные графическим процессором симуляции молекулярной динамики с помощью Gromacs в Google Cloud, используя пакетное задание Slurm.
Что вам понадобится
2. Настройка
Для выполнения этого практического задания вам необходимо включить API Compute Engine и Cloud Build. Чтобы включить API Compute Engine и Cloud Build, откройте Cloud Shell и выполните следующие команды. Не забудьте заменить идентификатор вашего проекта на...
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Внимание: Если вы планируете использовать сторонний SSH (например, OpenSSH) для подключения к кластеру, обязательно добавьте SSH-ключ в свой профиль облачной идентификации с помощью входа в ОС. Подробнее о добавлении SSH-ключей в профиль облачной идентификации можно узнать здесь .
3. [НЕОБЯЗАТЕЛЬНО] Создайте образ виртуальной машины GCP с помощью Gromacs
Для этого практического занятия мы предоставили вам предварительно собранный образ, поскольку процесс сборки Gromacs и всех его зависимостей может занять до 2 часов. Если вы хотите использовать этот предварительно собранный образ, чтобы сэкономить время, можете перейти к следующему разделу.
При запуске исследовательских приложений в Google Cloud существует множество вариантов установки и развертывания приложения. В этом разделе практического занятия вы создадите образ виртуальной машины, основанный на образе slurm-gcp (CentOS7). В процессе создания будут установлены компилятор, все зависимости Gromacs и сам Gromacs.
Конвейер сборки Gromacs Cloud Build в репозитории RCC Apps содержит необходимые инструкции для установки Gromacs. В процессе установки используется Packer для развертывания виртуальной машины, которая устанавливает Spack , который, в свою очередь, устанавливает компилятор GCC@9.2.0 и Gromacs@2021.2 с включенным ускорением GPU.
- Откройте Cloud Shell в GCP.
- Клонируйте репозиторий FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Создайте образ с помощью Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Вы можете проверить статус процесса сборки на панели управления Google Cloud Build.
Процесс сборки может занять до двух часов. Для его ускорения можно изменить схему файла конфигурации сборки , указав тип машины, чтобы повысить производительность сборки. Это можно сделать с помощью переменной сборки _MACHINE_TYPE. Например:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
После завершения сборки в вашем проекте Google Cloud появится образ виртуальной машины, который вы сможете использовать для развертывания кластера.
4. Разверните кластер высокопроизводительных вычислений с автоматическим масштабированием с помощью Terraform.
В этом разделе вы будете использовать Terraform для развертывания кластера высокопроизводительных вычислений с автоматическим масштабированием и установленным планировщиком заданий Slurm. Этот кластер будет развернут с вычислительными узлами, каждый из которых имеет 8 виртуальных процессоров и 1 графический процессор Nvidia® Tesla V100.
- Откройте Cloud Shell в GCP.
- Клонируйте репозиторий FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Перейдите в каталог gromacs terraform:
cd ~/rcc-apps/gromacs/tf/slurm
- Создайте и проверьте план Terraform. Установите переменные среды
GMX_NAME,GMX_PROJECTиGMX_ZONEчтобы указать имя вашего кластера, ваш проект GCP и зону, в которую вы хотите выполнить развертывание. Если у вас возникли вопросы, ознакомьтесь с примечанием ниже.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Если вы создали собственный образ виртуальной машины в предыдущем разделе этого практического задания, вам также потребуется установить переменную среды GMX_IMAGE.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Создайте план с помощью команды `make`, которая выполнит
terraform init && terraform plan.
make plan
- Разверните кластер. Процесс настройки занимает всего несколько минут, поскольку Gromacs и его зависимости уже предустановлены в вашем кластере.
make apply
- Подключитесь по SSH к узлу авторизации , созданному на предыдущем шаге. Вы можете увидеть этот узел на предыдущем шаге (вероятно, он называется gromacs-login0 ) . Это можно сделать, нажав кнопку SSH рядом со списком экземпляров виртуальных машин в меню консоли: Compute Engine -> Экземпляр виртуальной машины.
Дополнительная опция: Эта пара команд gcloud определит имя узла для входа и подключится к нему по SSH:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- После подключения к узлу авторизации, чтобы проверить настройку кластера, убедитесь, что Gromacs установлен.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Убедитесь, что
/opt/share/gromacsсодержит перечисленные ниже данные.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Запустите тест производительности benchRIB.
Gromacs — это исследовательское программное обеспечение, используемое для моделирования молекулярной динамики и расчета молекулярных структур с учетом ограничений минимизации энергии. Представленные в образе виртуальной машины задачи для этой лабораторной работы сосредоточены на молекулярной динамике, то есть на эволюции систем атомов.
В молекулярной динамике положения, скорости и ускорения атомов моделируются с использованием законов движения Ньютона:
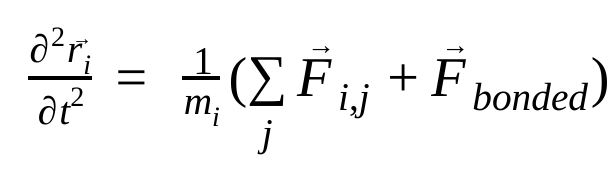
где, 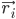 — это положение атома i , t — время.
— это положение атома i , t — время. 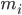 — это масса атома i, и
— это масса атома i, и 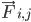 — это сила, возникающая в результате взаимодействия атома i с атомом j , и
— это сила, возникающая в результате взаимодействия атома i с атомом j , и 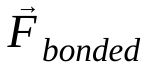 Силы обусловлены взаимодействиями между атомами. Зная температуру, давление, положения и скорости атомов, рассчитываются эти силы, и система численно интегрируется для получения новых скоростей и положений атомов. Этот процесс повторяется для моделирования молекулярной динамики в течение заданного периода времени.
Силы обусловлены взаимодействиями между атомами. Зная температуру, давление, положения и скорости атомов, рассчитываются эти силы, и система численно интегрируется для получения новых скоростей и положений атомов. Этот процесс повторяется для моделирования молекулярной динамики в течение заданного периода времени.
Образ Gromacs (созданный вами или предоставленный вами) содержит три бенчмарка.
- benchMEM
- benchRIB
- benchPEP
Эти тестовые примеры взяты из набора бесплатных тестов Gromacs, созданных доктором Кутцнером, и представляют собой стандартный набор симуляций молекулярной динамики в переходных процессах. Каждый тест отличается количеством атомов и продолжительностью симуляции. Соответствующие конфигурации для каждой симуляции приведены в таблице ниже.
Метрика / Эталон | benchMEM | benchRIB | benchPEP |
Количество атомов | 81,743 | 2 136 412 | 12,495,503 |
Размер системы / нм | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50.0 x 50.0 x 50.0 |
Шаг по времени / fs | 2 | 4 | 2 |
Радиусы отсечки / нм | 1 | 1 | 1.2 |
Шаг сетки PME / нм | 0,12 | 0,135 | 0,16 |
Для запуска теста производительности вам потребуется отправить пакетное задание Slurm. По умолчанию предоставленный пакетный скрипт запускает тест производительности benchRIB. Входные файлы, определяющие конфигурации для предоставленных тестов, включены в образ виртуальной машины Gromacs в каталоге /opt/share/gromacs . Кроме того, пример bash-скрипта для запуска Gromacs доступен в каталоге /opt/share .
Для выполнения этого раздела необходимо подключиться по SSH к узлу входа в кластер.
- Отправьте пакетное задание, используя команду sbatch.
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Это поставит задание в очередь на выполнение, и Slurm выделит вам вычислительный узел. При запуске sinfo вы увидите, что вычислительный узел находится в состоянии alloc# , что означает, что вычислительный узел выделен для вашего задания, но находится в процессе выделения. После запуска задания узел будет переведен в состояние alloc .
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Дождитесь завершения задания. Стандартный бенчмарк (benchRIB) моделирует около 8 миллионов атомов и настроен на выполнение 5000 временных шагов (с 4 временными шагами/секундой), что занимает около 6 минут. Вы можете отслеживать статус задания с помощью этой команды:
watch squeue
Когда ваше задание будет удалено из очереди, вы можете выйти, нажав Ctrl+C.
- После завершения работы вы должны увидеть каталог
run/содержащий результаты моделирования (вrun/MEM) и лог-файл в текущем каталоге под названиемgromacs.out. Каталогrun/MEMсодержит два файла:ener.edrиmd.log. Файлener.edrхранит энергии системы, температуру, давление, размер ячейки, плотность и вириалы в переносимом бинарном формате. Как следует из расширения файла, файлmd.logсодержит логи моделирования Gromacs и включает информацию о производительности моделирования, а также информацию из решателей PME и PME. Содержимое gromacs.out содержит стандартный вывод командыgmx mdrunи должно выглядеть примерно так, как показано ниже.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Поздравляем!
В этом практическом задании вы создали масштабируемый облачный высокопроизводительный вычислительный кластер и запустили моделирование молекулярной динамики с ускорением на графическом процессоре с помощью Gromacs в Google Cloud!
Уборка
Чтобы избежать списания средств с вашего аккаунта Google Cloud за ресурсы, использованные в этом практическом задании:
РЕКОМЕНДУЕТСЯ: Удалить кластер высокопроизводительных вычислений с помощью Terraform.
- Откройте CloudShell и перейдите в каталог примеров
gromacs/tf/slurm
cd ~/rcc-apps/gromacs/tf/slurm
- Выполните команду make destroy, чтобы удалить все ресурсы.
make destroy
ИЛИ : Удалить проект (наиболее эффективный и разрушительный вариант).
Самый простой способ избежать выставления счетов — удалить проект, созданный вами для практического занятия.
Внимание : удаление проекта влечет за собой следующие последствия:
- Все данные в проекте удаляются. Если вы использовали существующий проект для этой практической работы, то при его удалении вы также удалите всю остальную работу, выполненную в этом проекте.
- Пользовательские идентификаторы проектов теряются. При создании этого проекта вы могли создать пользовательский идентификатор проекта, который хотите использовать в будущем. Чтобы сохранить URL-адреса, использующие этот идентификатор проекта, например, URL-адрес appspot.com , удалите выбранные ресурсы внутри проекта, а не удаляйте весь проект.
Если вы планируете изучать несколько практических заданий и быстрых стартов, повторное использование проектов поможет вам избежать превышения лимитов квот на проекты.
- В консоли Cloud перейдите на страницу « Управление ресурсами ».
- В списке проектов выберите проект, который хотите удалить, и нажмите «Удалить».
 .
. - В диалоговом окне введите идентификатор проекта, а затем нажмите «Завершить» , чтобы удалить проект.