
1. บทนำ
อัปเดตล่าสุด: 25-04-2022
สิ่งที่คุณจะสร้าง
ใน Codelab นี้ คุณจะได้ทำให้ใช้งานได้คลัสเตอร์การประมวลผลประสิทธิภาพสูง (HPC) ที่ปรับขนาดอัตโนมัติใน Google Cloud การทำให้ใช้งานได้ Terraform จะสร้างคลัสเตอร์นี้โดยติดตั้ง Gromacs ผ่าน Spack คลัสเตอร์จะได้รับการจัดการด้วยตัวกำหนดเวลางาน Slurm เมื่อสร้างคลัสเตอร์แล้ว คุณจะเรียกใช้การเปรียบเทียบ benchMEM, benchPEP หรือ benchRIB
สิ่งที่คุณจะได้เรียนรู้
- วิธีการติดตั้งใช้งานคลัสเตอร์ HPC ที่รองรับตัวจัดกำหนดการงาน Slurm
- วิธีเรียกใช้การจำลองโมเลกุลไดนามิกที่เร่งด้วย GPU ด้วย Gromacs บน Google Cloud โดยใช้ชื่องานแบบกลุ่มของ Slurm
สิ่งที่คุณจะต้องมี
2. ตั้งค่า
คุณต้องเปิดใช้ Compute Engine API และ Cloud Build API เพื่อทำ Codelab นี้ให้เสร็จสมบูรณ์ หากต้องการเปิดใช้ Compute Engine และ Cloud Build API ให้เปิด Cloud Shell แล้วเรียกใช้คำสั่งต่อไปนี้ อย่าลืมแทนที่ project-id ด้วย
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
ข้อควรระวัง: หากวางแผนที่จะใช้ SSH ของบุคคลที่สาม (เช่น OpenSSH) เพื่อเชื่อมต่อกับคลัสเตอร์ โปรดตรวจสอบว่าได้แนบคีย์ SSH กับโปรไฟล์ Cloud Identity โดยใช้การเข้าสู่ระบบปฏิบัติการ ดูข้อมูลเพิ่มเติมเกี่ยวกับการเพิ่มคีย์ SSH ลงในโปรไฟล์ Cloud Identity
3. [ไม่บังคับ] สร้างอิมเมจ VM ของ GCP ด้วย Gromacs
สำหรับ Codelab นี้ เราได้จัดเตรียมอิมเมจที่สร้างไว้ล่วงหน้าให้คุณแล้ว เนื่องจากกระบวนการบิลด์อาจใช้เวลาถึง 2 ชั่วโมงในการติดตั้ง Gromacs และทรัพยากร Dependency ทั้งหมด หากต้องการใช้รูปภาพที่สร้างไว้ล่วงหน้านี้เพื่อประหยัดเวลา ให้ข้ามไปยังส่วนถัดไป
เมื่อเรียกใช้แอปพลิเคชันการวิจัยใน Google Cloud คุณจะมีตัวเลือกมากมายในการติดตั้งและทําให้แอปพลิเคชันใช้งานได้ ในส่วนนี้ของ Codelab คุณจะสร้างอิมเมจเครื่องเสมือนที่สร้างขึ้นจากอิมเมจ VM ของ slurm-gcp (CentOS7) ในระหว่างกระบวนการสร้าง ระบบจะติดตั้งคอมไพเลอร์ การขึ้นต่อกันทั้งหมดของ Gromacs และ Gromacs
ไปป์ไลน์การสร้าง Gromacs Cloud ในที่เก็บแอป RCC จะห่อหุ้มวิธีการที่จำเป็นสำหรับการติดตั้ง Gromacs กระบวนการติดตั้งใช้ Packer เพื่อติดตั้ง VM ที่ติดตั้ง Spack ซึ่งจะติดตั้งคอมไพเลอร์ GCC@9.2.0 และ Gromacs@2021.2 โดยเปิดใช้การเร่งด้วย GPU
- เปิด Cloud Shell ใน GCP
- โคลนที่เก็บ FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- สร้างอิมเมจโดยใช้ Google Cloud Build
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
คุณตรวจสอบสถานะของกระบวนการบิลด์ได้ที่แดชบอร์ด Google Cloud Build
กระบวนการบิลด์อาจใช้เวลาถึง 2 ชั่วโมง หากต้องการเร่งกระบวนการนี้ คุณอาจพิจารณาแก้ไขสคีมาไฟล์การกำหนดค่าบิลด์เพื่อเปลี่ยนประเภทเครื่องเพื่อปรับปรุงประสิทธิภาพการบิลด์ ซึ่งทำได้โดยใช้ตัวแปรการสร้าง _MACHINE_TYPE เช่น
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
เมื่อบิลด์เสร็จสมบูรณ์แล้ว คุณจะมีอิมเมจ VM ในโปรเจ็กต์ที่อยู่ในระบบคลาวด์ Google Cloud ซึ่งใช้เพื่อทำให้ใช้งานได้คลัสเตอร์ได้
4. ติดตั้งใช้งานคลัสเตอร์ HPC ที่ปรับขนาดอัตโนมัติด้วย Terraform
ในส่วนนี้ คุณจะใช้ Terraform เพื่อทำให้คลัสเตอร์ HPC ที่ปรับขนาดอัตโนมัติใช้งานได้ โดยติดตั้งตัวจัดกำหนดการงาน Slurm ไว้ด้วย คลัสเตอร์นี้จะได้รับการติดตั้งใช้งานด้วยโหนดการคำนวณซึ่งแต่ละโหนดมี vCPU 8 รายการและ GPU Nvidia® Tesla V100 1 รายการ
- เปิด Cloud Shell ใน GCP
- โคลนที่เก็บ FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- เปลี่ยนเป็นไดเรกทอรี Terraform ของ Gromacs โดยใช้คำสั่งต่อไปนี้
cd ~/rcc-apps/gromacs/tf/slurm
- สร้างและตรวจสอบแผน Terraform ตั้งค่าตัวแปรสภาพแวดล้อม
GMX_NAME,GMX_PROJECTและGMX_ZONEเพื่อระบุชื่อคลัสเตอร์ โปรเจ็กต์ GCP และโซนที่คุณต้องการติดตั้งใช้งาน โปรดอ่านหมายเหตุด้านล่างหากไม่แน่ใจ
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- หากคุณสร้างอิมเมจ VM ของตัวเองในส่วนก่อนหน้าของ Codelab นี้ คุณจะต้องตั้งค่าตัวแปรสภาพแวดล้อม GMX_IMAGE ด้วย
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- สร้างแผนด้วยคำสั่ง make ซึ่งจะเรียกใช้
terraform init && terraform plan
make plan
- ทำให้คลัสเตอร์ใช้งานได้ กระบวนการตั้งค่าใช้เวลาเพียงไม่กี่นาที เนื่องจาก Gromacs และการขึ้นต่อกันจะได้รับการติดตั้งไว้ล่วงหน้าในคลัสเตอร์
make apply
- SSH ไปยังโหนด login ที่สร้างไว้ในขั้นตอนก่อนหน้า คุณจะเห็นโหนดนี้ในขั้นตอนก่อนหน้า (อาจมีชื่อว่า gromacs-login0) คุณทำได้โดยคลิกปุ่ม SSH ข้างรายการอินสแตนซ์ VM ในรายการในเมนูคอนโซล Compute Engine -> อินสแตนซ์ VM
ตัวเลือก: คำสั่ง gcloud 2 รายการนี้จะค้นหาชื่อโหนดการเข้าสู่ระบบและ SSH เข้าไป
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- เมื่อเชื่อมต่อกับโหนดการเข้าสู่ระบบแล้ว ให้ตรวจสอบว่าได้ติดตั้ง Gromacs แล้วเพื่อยืนยันการตั้งค่าคลัสเตอร์
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- ตรวจสอบว่า
/opt/share/gromacsมีเนื้อหาตามที่ระบุไว้ด้านล่าง
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. เรียกใช้การเปรียบเทียบ benchRIB
Gromacs เป็นซอฟต์แวร์วิจัยที่ใช้ในการจำลองพลศาสตร์ระดับโมเลกุลและคำนวณโครงสร้างโมเลกุลภายใต้ข้อจำกัดของการลดพลังงาน เกณฑ์มาตรฐานที่ระบุไว้ในอิมเมจ VM สำหรับ Codelab นี้มุ่งเน้นไปที่พลศาสตร์โมเลกุล ซึ่งเป็นวิวัฒนาการของระบบอะตอม
ในการพลศาสตร์โมเลกุล ระบบจะจำลองตำแหน่ง ความเร็ว และความเร่งของอะตอมโดยใช้กฎการเคลื่อนที่ของนิวตัน ดังนี้
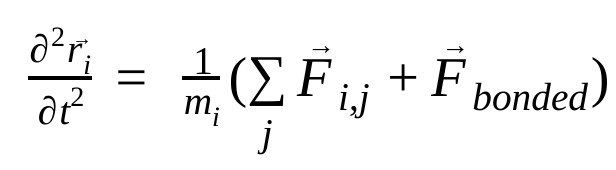
โดยที่ 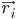 คือตำแหน่งของอะตอม i, t คือเวลา,
คือตำแหน่งของอะตอม i, t คือเวลา, 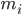 คือมวลของอะตอม i และ
คือมวลของอะตอม i และ 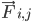 คือแรงที่ไม่ใช่พันธะบนอะตอม i เนื่องจากอะตอม j และ
คือแรงที่ไม่ใช่พันธะบนอะตอม i เนื่องจากอะตอม j และ 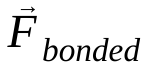 คือแรงเนื่องจากการโต้ตอบแบบพันธะ เมื่อทราบอุณหภูมิ ความดัน ตำแหน่งอะตอม และความเร็วอะตอมแล้ว ระบบจะคำนวณแรงและรวมระบบแบบตัวเลขเพื่อหาความเร็วและตำแหน่งใหม่ของอะตอม กระบวนการนี้จะทำซ้ำเพื่อจำลองพลศาสตร์ระดับโมเลกุลในช่วงระยะเวลาหนึ่ง
คือแรงเนื่องจากการโต้ตอบแบบพันธะ เมื่อทราบอุณหภูมิ ความดัน ตำแหน่งอะตอม และความเร็วอะตอมแล้ว ระบบจะคำนวณแรงและรวมระบบแบบตัวเลขเพื่อหาความเร็วและตำแหน่งใหม่ของอะตอม กระบวนการนี้จะทำซ้ำเพื่อจำลองพลศาสตร์ระดับโมเลกุลในช่วงระยะเวลาหนึ่ง
รูปภาพ Gromacs (ไม่ว่าจะเป็นรูปภาพที่คุณสร้างขึ้นหรือรูปภาพที่ให้ไว้) จะมาพร้อมกับการเปรียบเทียบ 3 รายการ
- benchMEM
- benchRIB
- benchPEP
เกณฑ์เปรียบเทียบเหล่านี้มาจากชุดเกณฑ์เปรียบเทียบ Gromacs ฟรีของดร. Kutzner และเป็นชุดมาตรฐานของการจำลองโมเลกุลแบบไดนามิกชั่วคราว การเปรียบเทียบแต่ละรายการจะแตกต่างกันในด้านจำนวนอะตอมและระยะเวลาของการจำลอง การกำหนดค่าที่เกี่ยวข้องสำหรับการจำลองแต่ละครั้งจะแสดงในตารางด้านล่าง
เมตริก / การเปรียบเทียบ | benchMEM | benchRIB | benchPEP |
จำนวน Atoms | 81,743 | 2,136,412 | 12,495,503 |
ขนาดระบบ / nm | 10.8 x 10.2 x 9.6 | 31.2 x 31.2 x 31.2 | 50.0 x 50.0 x 50.0 |
Time Step / fs | 2 | 4 | 2 |
รัศมีการตัด / nm | 1 | 1 | 1.2 |
ระยะห่างของตาราง PME / nm | 0.12 | 0.135 | 0.16 |
หากต้องการเรียกใช้การทดสอบเปรียบเทียบ คุณจะต้องส่งงานแบบกลุ่มของ Slurm โดยค่าเริ่มต้น สคริปต์ชุดที่ระบุจะเรียกใช้การเปรียบเทียบ benchRIB เด็คอินพุตที่กำหนดค่าสำหรับการเปรียบเทียบที่ระบุจะรวมอยู่ในอิมเมจ VM ของ Gromacs ในส่วน /opt/share/gromacs นอกจากนี้ ยังมีตัวอย่างสคริปต์ Bash สำหรับเรียกใช้ Gromacs อยู่ในส่วน /opt/share
สำหรับส่วนนี้ คุณต้อง SSH ไปยังโหนด login ของคลัสเตอร์
- ส่งงานแบบกลุ่มโดยใช้คำสั่ง sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
ซึ่งจะจัดคิวงานเพื่อดำเนินการและ Slurm จะจัดสรรโหนดการคำนวณให้คุณ เมื่อเรียกใช้ sinfo คุณจะเห็นว่าโหนดการคำนวณอยู่ในสถานะ alloc# ซึ่งหมายความว่าระบบได้จัดสรรโหนดการคำนวณสำหรับงานของคุณแล้ว แต่กำลังอยู่ในกระบวนการจัดสรร เมื่องานทำงานแล้ว ระบบจะตั้งค่าโหนดเป็นสถานะ alloc
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
รอให้งานเสร็จสมบูรณ์ การเปรียบเทียบเริ่มต้น (benchRIB) จะจำลองอะตอมประมาณ 8 ล้านอะตอม และได้รับการกำหนดค่าให้ทำงาน 5,000 ขั้นตอนเวลา (โดยมี 4 ขั้นตอนเวลา/fs) และใช้เวลาประมาณ 6 นาทีจึงจะเสร็จสมบูรณ์ คุณตรวจสอบสถานะของงานได้ด้วยคำสั่งนี้
watch squeue
เมื่องานของคุณไม่อยู่ในคิวแล้ว คุณจะออกได้ด้วยการกด Ctrl-C
- เมื่องานเสร็จสมบูรณ์แล้ว คุณจะเห็นไดเรกทอรีชื่อ
run/ซึ่งมีเอาต์พุตการจำลอง (ในrun/MEM) และไฟล์บันทึกในไดเรกทอรีปัจจุบันชื่อgromacs.outไดเรกทอรีrun/MEMมีไฟล์ 2 ไฟล์ ได้แก่ener.edrและmd.logener.edrไฟล์จะจัดเก็บพลังงาน อุณหภูมิ ความดัน ขนาดกล่อง ความหนาแน่น และวิริอัลของระบบในรูปแบบไบนารีแบบพกพา ตามที่ส่วนขยายแนะนำmd.logไฟล์นี้มีบันทึกสำหรับการจำลอง Gromacs และมีข้อมูลเกี่ยวกับประสิทธิภาพการจำลองนอกเหนือจากข้อมูลการบันทึกจากตัวแก้ปัญหาอนุภาค-อนุภาคและ PME เนื้อหาของ gromacs.out มีเอาต์พุตมาตรฐานจากgmx mdrunควรมีลักษณะคล้ายกับที่แสดงด้านล่าง
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. ขอแสดงความยินดี
ใน Codelab นี้ คุณได้สร้างคลัสเตอร์ HPC ที่ดำเนินการบนระบบคลาวด์ที่ปรับขนาดอัตโนมัติ และเรียกใช้การจำลองพลศาสตร์โมเลกุลที่เร่งด้วย GPU ด้วย Gromacs ใน Google Cloud
การล้างข้อมูล
โปรดดำเนินการดังนี้เพื่อเลี่ยงไม่ให้เกิดการเรียกเก็บเงินกับบัญชี Google Cloud สำหรับทรัพยากรที่ใช้ใน Codelab นี้
แนะนำ: ลบคลัสเตอร์ HPC ด้วย Terraform
- เปิด Cloud Shell แล้วไปที่ไดเรกทอรีตัวอย่าง
gromacs/tf/slurm
cd ~/rcc-apps/gromacs/tf/slurm
- เรียกใช้ make destroy เพื่อลบทรัพยากรทั้งหมด
make destroy
หรือลบโปรเจ็กต์ (มีประสิทธิภาพและทำลายล้างมากที่สุด)
วิธีที่ง่ายที่สุดในการยกเลิกการเรียกเก็บเงินคือการลบโปรเจ็กต์ที่คุณสร้างขึ้นสำหรับโค้ดแล็บ
ข้อควรระวัง: การลบโปรเจ็กต์จะมีผลดังต่อไปนี้
- ระบบจะลบทุกอย่างในโปรเจ็กต์ หากใช้โปรเจ็กต์ที่มีอยู่สำหรับ Codelab นี้ เมื่อลบโปรเจ็กต์ ระบบจะลบงานอื่นๆ ที่คุณทำในโปรเจ็กต์ด้วย
- รหัสโปรเจ็กต์ที่กำหนดเองจะหายไป เมื่อสร้างโปรเจ็กต์นี้ คุณอาจสร้างรหัสโปรเจ็กต์ที่กำหนดเองซึ่งต้องการใช้ในอนาคต หากต้องการเก็บ URL ที่ใช้รหัสโปรเจ็กต์ เช่น URL ของ appspot.com ให้ลบทรัพยากรที่เลือกภายในโปรเจ็กต์แทนการลบทั้งโปรเจ็กต์
หากวางแผนที่จะสำรวจ Codelab และการเริ่มต้นใช้งานด่วนหลายรายการ การนำโปรเจ็กต์กลับมาใช้ใหม่จะช่วยให้คุณไม่เกินขีดจำกัดโควต้าของโปรเจ็กต์
- ใน Cloud Console ให้ไปที่หน้าจัดการทรัพยากร ไปที่หน้าจัดการทรัพยากร
- ในรายการโปรเจ็กต์ ให้เลือกโปรเจ็กต์ที่ต้องการลบ แล้วคลิกลบ

- ในกล่องโต้ตอบ ให้พิมพ์รหัสโปรเจ็กต์ แล้วคลิกปิดเพื่อลบโปรเจ็กต์