
1. Giriş
Son güncelleme: 2022-04-25
Ne oluşturacaksınız?
Bu codelab'de, Google Cloud'da otomatik ölçeklendirmeli bir yüksek performanslı bilgi işlem (HPC) kümesi dağıtacaksınız.Bu küme, Spack aracılığıyla yüklenen Gromacs ile bir Terraform dağıtımı tarafından oluşturulur. Küme, Slurm işi planlayıcı ile yönetilir. Küme oluşturulduğunda benchMEM, benchPEP veya benchRIB kıyaslamalarını çalıştırırsınız.
Öğrenecekleriniz
- Slurm iş zamanlayıcısını destekleyen bir HPC kümesi dağıtma
- Slurm toplu işini kullanarak Google Cloud'da Gromacs ile GPU hızlandırmalı moleküler dinamik simülasyonları çalıştırma
İhtiyacınız olanlar
2. Kurulum
Bu codelab'i tamamlamak için Compute Engine ve Cloud Build API'lerini etkinleştirmeniz gerekir. Compute Engine ve Cloud Build API'lerini etkinleştirmek için Cloud Shell'inizi açın ve aşağıdaki komutları çalıştırın. Aşağıdaki
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Dikkat: Kümenize bağlanmak için üçüncü taraf SSH (ör. OpenSSH) kullanmayı planlıyorsanız OS girişi ile bulut kimliği profilinize bir SSH anahtarı eklediğinizden emin olun. Cloud Identity profilinize SSH anahtarları ekleme hakkında daha fazla bilgi edinin.
3. [İSTEĞE BAĞLI] Gromacs ile GCP sanal makine görüntüsü oluşturma
Bu codelab'de, Gromacs'ın ve tüm bağımlılıklarının yüklenmesi 2 saati bulabileceğinden sizin için önceden oluşturulmuş bir resim sağladık. Zaman kazanmak için bu önceden oluşturulmuş resmi kullanmak isterseniz bir sonraki bölüme geçebilirsiniz.
Google Cloud'da araştırma uygulamaları çalıştırırken uygulamanızı yüklemek ve dağıtmak için birçok seçenek vardır. Bu codelab bölümünde, slurm-gcp (CentOS7) sanal makine görüntüsünün üzerine oluşturulan bir sanal makine görüntüsü oluşturacaksınız. Oluşturma işlemi sırasında derleyici, Gromacs'in tüm bağımlılıkları ve Gromacs yüklenir.
RCC Apps deposundaki Gromacs Cloud Build ardışık düzeni, Gromacs'ı yüklemek için gerekli talimatları kapsar. Yükleme işlemi, Packer'ı kullanarak Spack'i yükleyen bir sanal makine dağıtır. Spack ise GCC@9.2.0 derleyicisini ve GPU hızlandırması etkinleştirilmiş Gromacs@2021.2'yi yükler.
- GCP'de Cloud Shell'inizi açın.
- FluidNumerics/rcc-apps deposunu klonlayın.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Google Cloud Build'i kullanarak görüntüyü oluşturun.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Derleme işleminizin durumunu Google Cloud Build kontrol panelinden kontrol edebilirsiniz.
Derleme işlemi iki saat sürebilir. Bu işlemi hızlandırmak için derleme performansını artırmak amacıyla makine türünü değiştirmek üzere derleme yapılandırma dosyası şemanızı değiştirebilirsiniz. Bu işlem, _MACHINE_TYPE derleme değişkeni kullanılarak yapılabilir. Örneğin:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Derlemeniz tamamlandığında, kümenizi dağıtmak için kullanabileceğiniz bir sanal makine görüntüsü Google Cloud projenizde kullanılabilir.
4. Terraform ile otomatik ölçeklendirmeli HPC kümesi dağıtma
Bu bölümde, Slurm iş zamanlayıcısının yüklü olduğu otomatik ölçeklendirmeli bir HPC kümesi dağıtmak için Terraform'u kullanacaksınız. Bu küme, her biri 8 sanal CPU ve 1 Nvidia® Tesla V100 GPU'ya sahip işlem düğümleriyle dağıtılır.
- GCP'de Cloud Shell'inizi açın.
- FluidNumerics/rcc-apps deposunu klonlayın.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Gromacs terraform dizinine geçin:
cd ~/rcc-apps/gromacs/tf/slurm
- Terraform planı oluşturma ve inceleme Kümenizin adını, GCP projenizi ve dağıtım yapmak istediğiniz bölgeyi belirtmek için
GMX_NAME,GMX_PROJECTveGMX_ZONEortam değişkenlerini ayarlayın. Emin değilseniz lütfen aşağıdaki notu inceleyin.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Bu codelab'in önceki bölümünde kendi sanal makine resminizi oluşturduysanız GMX_IMAGE ortam değişkenini de ayarlamanız gerekir.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Planı,
terraform init && terraform plankomutunu çalıştıracak olan make komutuyla oluşturun.
make plan
- Kümeyi dağıtın. Gromacs ve bağımlılıkları kümenize önceden yüklenmiş olarak geldiğinden kurulum işlemi yalnızca birkaç dakika sürer.
make apply
- Önceki adımda oluşturulan login düğümüne SSH uygulayın. Bu düğümü önceki adımda (büyük olasılıkla gromacs-login0 olarak adlandırılır) görebilirsiniz. Bunu, konsol menü öğesi Compute Engine -> VM instance'da (Compute Engine -> Sanal makine örneği) VM örnekleri listesinin yanındaki SSH düğmesini tıklayarak yapabilirsiniz.
Seçenek: Bu gcloud komutları çifti, giriş düğümü adını bulur ve SSH ile bağlanır:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Giriş düğümüne bağlandıktan sonra küme kurulumunuzu doğrulamak için Gromacs'in yüklendiğini kontrol edin.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
/opt/share/gromacsiçinde aşağıda listelenen içeriklerin bulunduğunu doğrulayın.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. benchRIB karşılaştırmasını çalıştırma
Gromacs, moleküler dinamikleri simüle etmek ve moleküler yapıları enerji minimizasyonu kısıtlamaları altında hesaplamak için kullanılan bir araştırma yazılımıdır. Bu codelab'in sanal makine görüntüsünde sağlanan karşılaştırma ölçütleri, atom sistemlerinin evrimi olan moleküler dinamiklere odaklanır.
Moleküler dinamikte, atomların konumları, hızları ve ivmeleri Newton'un hareket yasaları kullanılarak simüle edilir :
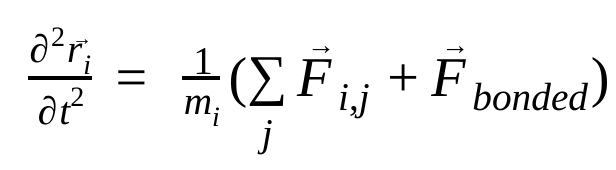
Burada 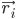 , i atomunun konumunu, t zamanı,
, i atomunun konumunu, t zamanı, 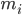 , i atomunun kütlesini,
, i atomunun kütlesini, 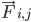 , j atomu nedeniyle i atomu üzerindeki bağsız kuvveti ve
, j atomu nedeniyle i atomu üzerindeki bağsız kuvveti ve 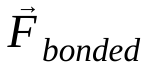 , bağlı etkileşimlerden kaynaklanan kuvvetleri ifade eder. Sıcaklık, basınç, atom konumları ve atom hızları göz önüne alındığında kuvvetler hesaplanır ve yeni atom hızları ile konumları elde etmek için sistem sayısal olarak entegre edilir. Bu işlem, belirli bir süre boyunca moleküler dinamikleri simüle etmek için tekrarlanır.
, bağlı etkileşimlerden kaynaklanan kuvvetleri ifade eder. Sıcaklık, basınç, atom konumları ve atom hızları göz önüne alındığında kuvvetler hesaplanır ve yeni atom hızları ile konumları elde etmek için sistem sayısal olarak entegre edilir. Bu işlem, belirli bir süre boyunca moleküler dinamikleri simüle etmek için tekrarlanır.
Gromacs görüntüsü (oluşturduğunuz veya sağlanan) üç karşılaştırma testiyle birlikte gelir.
- benchMEM
- benchRIB
- benchPEP
Bu karşılaştırma testleri, Dr. Kutzner'ın Ücretsiz Gromacs Karşılaştırma Testleri 'nden alınmıştır ve geçici moleküler dinamik simülasyonlarının standart bir kümesidir. Her karşılaştırma, atom sayısı ve simülasyon süresi bakımından farklılık gösterir. Her simülasyonla ilgili yapılandırmalar aşağıdaki tabloda verilmiştir.
Metrik / Karşılaştırma | benchMEM | benchRIB | benchPEP |
Atom Sayısı | 81.743 | 2.136.412 | 12.495.503 |
Sistem Boyutu / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50,0 x 50,0 x 50,0 |
Zaman Adımı / fs | 2 | 4 | 2 |
Kesme yarıçapları / nm | 1 | 1 | 1,2 |
PME ızgara aralığı / nm | 0,12 | 0,135 | 0,16 |
Karşılaştırmayı çalıştırmak için bir Slurm toplu işi göndereceksiniz. Varsayılan olarak, sağlanan toplu komut dosyası benchRIB karşılaştırmasını çalıştırır. Sağlanan kıyaslamaların yapılandırmalarını tanımlayan giriş desteleri, /opt/share/gromacs altında Gromacs VM görüntüsüne dahil edilir. Ayrıca, Gromacs'ı çalıştırmak için örnek bir bash komut dosyası /opt/share altında mevcuttur.
Bu bölüm için kümenin login düğümüne SSH yapmanız gerekir.
- sbatch komutunu kullanarak toplu iş gönderme
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Bu işlem, işi yürütülmek üzere sıraya alır ve Slurm sizin için bir işlem düğümü sağlar. sinfo komutunu çalıştırdığınızda bir işlem düğümünün alloc# durumunda olduğunu görürsünüz. Bu, işlem düğümünün işiniz için ayrıldığı ancak henüz sağlama sürecinde olduğu anlamına gelir. İşiniz çalışmaya başladığında düğüm alloc durumuna ayarlanır.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
İşin tamamlanmasını bekleyin. Varsayılan karşılaştırma (benchRIB) yaklaşık 8 milyon atomu simüle eder ve 5.000 zaman adımı (4 zaman adımı/fs) çalıştıracak şekilde yapılandırılmıştır. Tamamlanması yaklaşık 6 dakika sürer. Aşağıdaki komutla işinizin durumunu izleyebilirsiniz:
watch squeue
İşiniz kuyruktan çıktığında ctrl-C ile çıkabilirsiniz.
- İş tamamlandığında, geçerli dizininizde
run/adlı bir dizin (run/MEMaltında simülasyon çıkışını içerir) vegromacs.outadlı bir günlük dosyası görürsünüz.run/MEMdiziniener.edrvemd.logolmak üzere iki dosya içerir.ener.edrdosyası, sistemin enerjilerini, sıcaklığını, basıncını, kutu boyutunu, yoğunluğunu ve viriyallerini taşınabilir bir ikili biçimde depolar. Uzantının da belirttiği gibi,md.logdosyası Gromacs simülasyonunun günlüklerini içerir ve parçacık-parçacık ve PME çözücülerinden gelen günlük bilgilerinin yanı sıra simülasyon performansı hakkında bilgiler de içerir. gromacs.out dosyasının içeriği,gmx mdrunkomutunun standart çıkışını içerir ve aşağıda gösterilene benzer olmalıdır.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Tebrikler
Bu codelab'de, otomatik ölçeklendirmeli, bulutta yerel bir HPC kümesi oluşturdunuz ve Google Cloud'da Gromacs ile GPU hızlandırmalı bir moleküler dinamik simülasyonu çalıştırdınız.
Temizleme
Bu codelab'de kullanılan kaynaklar için Google Cloud hesabınızın ücretlendirilmesini istemiyorsanız:
ÖNERİLEN: HPC kümesini Terraform ile silme
- Cloud Shell'inizi açın ve
gromacs/tf/slurmörnek dizinine gidin.
cd ~/rcc-apps/gromacs/tf/slurm
- Tüm kaynakları silmek için make destroy komutunu çalıştırın.
make destroy
VEYA, projeyi silin (en etkili ve yıkıcı yöntem)
Faturalandırılmanın önüne geçmenin en kolay yolu, codelab için oluşturduğunuz projeyi silmektir.
Dikkat: Bir projeyi silmenin aşağıdaki etkileri olur:
- Projedeki her şey silinir. Bu codelab için mevcut bir proje kullandıysanız projeyi sildiğinizde projede yaptığınız diğer tüm çalışmalar da silinir.
- Özel proje kimlikleri kaybolur. Bu projeyi oluştururken gelecekte kullanmak istediğiniz özel bir proje kimliği oluşturmuş olabilirsiniz. Proje kimliğini kullanan URL'leri (ör. appspot.com URL'si) korumak için projenin tamamını silmek yerine proje içindeki seçili kaynakları silin.
Birden fazla codelab ve hızlı başlangıç kılavuzu incelemeyi planlıyorsanız projeleri yeniden kullanmak proje kotası sınırlarını aşmanızı önleyebilir.
- Cloud Console'da Kaynakları yönetin sayfasına gidin. Kaynakları yönetin sayfasına gidin
- Proje listesinde, silmek istediğiniz projeyi seçin ve Sil 'i
 tıklayın.
tıklayın. - İletişim kutusunda proje kimliğini yazın ve projeyi silmek için Kapat'ı tıklayın.