
1. Giới thiệu
Lần cập nhật gần đây nhất: ngày 25 tháng 4 năm 2022
Sản phẩm bạn sẽ tạo ra
Trong lớp học lập trình này, bạn sẽ triển khai một cụm Điện toán hiệu suất cao (HPC) tự động cấp tài nguyên bổ sung trên Google Cloud.Việc triển khai Terraform sẽ tạo cụm này bằng Gromacs được cài đặt thông qua Spack. Cụm sẽ được quản lý bằng trình lập lịch biểu lệnh Slurm. Khi tạo cụm, bạn sẽ chạy các phép đo điểm chuẩn benchMEM, benchPEP hoặc benchRIB.
Kiến thức bạn sẽ học được
- Cách triển khai một cụm HPC hỗ trợ trình lập lịch công việc Slurm
- Cách chạy mô phỏng động lực học phân tử được tăng tốc bằng GPU bằng Gromacs trên Google Cloud bằng cách sử dụng một lô công việc Slurm
Những gì bạn cần
2. Thiết lập
Để hoàn tất lớp học lập trình này , bạn cần bật API Compute Engine và Cloud Build. Để bật API Compute Engine và Cloud Build, hãy mở Cloud Shell rồi chạy các lệnh sau. Đừng quên thay thế project-id bằng
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Thận trọng: Nếu bạn dự định sử dụng SSH của bên thứ ba (ví dụ: OpenSSH) để kết nối với cụm, hãy nhớ đính khoá SSH vào hồ sơ danh tính trên đám mây bằng tính năng Đăng nhập bằng hệ điều hành. Tìm hiểu thêm về cách thêm khoá SSH vào hồ sơ danh tính trên đám mây.
3. [KHÔNG BẮT BUỘC] Tạo một hình ảnh máy ảo GCP bằng Gromacs
Trong lớp học lập trình này, chúng tôi đã cung cấp một hình ảnh dựng sẵn cho bạn, vì quy trình xây dựng có thể mất đến 2 giờ để cài đặt Gromacs và tất cả các phần phụ thuộc của nó. Nếu muốn sử dụng hình ảnh được tạo sẵn này để tiết kiệm thời gian, bạn có thể bỏ qua và chuyển sang phần tiếp theo.
Khi chạy các ứng dụng nghiên cứu trên Google Cloud, bạn có nhiều lựa chọn để cài đặt và triển khai ứng dụng của mình. Trong phần này của lớp học lập trình, bạn sẽ tạo một hình ảnh máy ảo dựa trên hình ảnh máy ảo slurm-gcp (CentOS7). Trong quá trình tạo, trình biên dịch, tất cả các phần phụ thuộc của Gromacs và Gromacs sẽ được cài đặt.
Quy trình Gromacs Cloud Build trên kho lưu trữ RCC Apps bao gồm các hướng dẫn cần thiết để cài đặt Gromacs. Quy trình cài đặt sử dụng Packer để triển khai một VM cài đặt Spack. Spack sẽ cài đặt trình biên dịch GCC@9.2.0 và Gromacs@2021.2 khi đã bật tính năng tăng tốc GPU.
- Mở Cloud Shell trên GCP.
- Sao chép kho lưu trữ FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Tạo hình ảnh bằng Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Bạn có thể kiểm tra trạng thái của quy trình xây dựng tại trang tổng quan Google Cloud Build
Quá trình xây dựng có thể mất đến 2 giờ. Để đẩy nhanh quá trình này,bạn có thể cân nhắc sửa đổi giản đồ tệp cấu hình bản dựng để thay đổi loại máy nhằm cải thiện hiệu suất bản dựng. Bạn có thể thực hiện việc này bằng cách sử dụng biến bản dựng _MACHINE_TYPE. Ví dụ:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Sau khi bản dựng hoàn tất, bạn sẽ có một hình ảnh máy ảo trong dự án trên đám mây Google Cloud mà bạn có thể dùng để triển khai cụm.
4. Triển khai một cụm HPC tự động cấp tài nguyên bổ sung bằng Terraform
Trong phần này, bạn sẽ sử dụng Terraform để triển khai một cụm HPC tự động cấp tài nguyên bổ sung, có trình lập lịch công việc Slurm được cài đặt. Cụm này sẽ được triển khai với các nút tính toán, mỗi nút có 8 vCPU và 1 GPU Nvidia® Tesla V100.
- Mở Cloud Shell trên GCP.
- Sao chép kho lưu trữ FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Thay đổi thành thư mục gromacs terraform:
cd ~/rcc-apps/gromacs/tf/slurm
- Tạo và xem xét kế hoạch terraform. Đặt các biến môi trường
GMX_NAME,GMX_PROJECTvàGMX_ZONEđể chỉ định tên của cụm, dự án GCP và vùng mà bạn muốn triển khai. Vui lòng xem kỹ lưu ý bên dưới nếu bạn không chắc chắn
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Nếu đã tạo hình ảnh VM của riêng mình trong phần trước của lớp học lập trình này, bạn cũng cần đặt biến môi trường GMX_IMAGE
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Tạo kế hoạch bằng lệnh make, lệnh này sẽ chạy
terraform init && terraform plan.
make plan
- Triển khai cụm. Quá trình thiết lập chỉ mất vài phút vì Gromacs và các phần phụ thuộc của nó được cài đặt sẵn trên cụm của bạn.
make apply
- SSH đến nút login được tạo ở bước trước. Bạn có thể thấy nút này ở bước trước (có thể được gọi là gromacs-login0). Bạn có thể thực hiện việc này bằng cách nhấp vào nút SSH bên cạnh danh sách các thực thể máy ảo trong mục trong trình đơn Compute Engine -> Phiên bản máy ảo của bảng điều khiển.
Lựa chọn: Cặp lệnh gcloud này sẽ xác định tên nút đăng nhập và truy cập vào nút đó qua SSH:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Sau khi kết nối với nút đăng nhập, để xác minh chế độ thiết lập cụm, hãy kiểm tra xem Gromacs đã được cài đặt hay chưa
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Xác minh rằng
/opt/share/gromacscó nội dung được liệt kê bên dưới.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Chạy phép đo điểm chuẩn benchRIB
Gromacs là phần mềm nghiên cứu được dùng để mô phỏng động lực học phân tử và tính toán cấu trúc phân tử theo các quy tắc ràng buộc giảm thiểu năng lượng. Các điểm chuẩn được cung cấp trong hình ảnh máy ảo cho lớp học lập trình này tập trung vào động lực học phân tử, sự phát triển của các hệ thống nguyên tử.
Trong động lực học phân tử, vị trí, vận tốc và gia tốc của các nguyên tử được mô phỏng bằng cách sử dụng các định luật chuyển động của Newton :
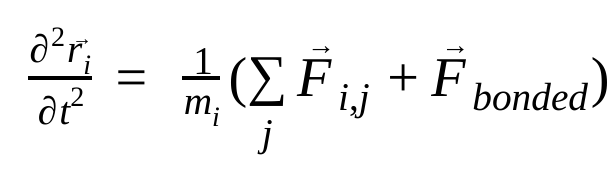
trong đó, 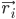 là vị trí của nguyên tử i, t là thời gian,
là vị trí của nguyên tử i, t là thời gian, 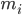 là khối lượng của nguyên tử i và
là khối lượng của nguyên tử i và 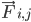 là lực không liên kết tác động lên nguyên tử i do nguyên tử j và
là lực không liên kết tác động lên nguyên tử i do nguyên tử j và 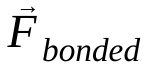 là các lực do tương tác liên kết. Với nhiệt độ, áp suất, vị trí nguyên tử và vận tốc nguyên tử, các lực được tính toán và hệ thống được tích hợp bằng số để thu được vận tốc và vị trí nguyên tử mới. Quá trình này được lặp lại để mô phỏng động lực học phân tử trong một khoảng thời gian nhất định.
là các lực do tương tác liên kết. Với nhiệt độ, áp suất, vị trí nguyên tử và vận tốc nguyên tử, các lực được tính toán và hệ thống được tích hợp bằng số để thu được vận tốc và vị trí nguyên tử mới. Quá trình này được lặp lại để mô phỏng động lực học phân tử trong một khoảng thời gian nhất định.
Hình ảnh Gromacs (do bạn tạo hoặc do chúng tôi cung cấp) đi kèm với 3 điểm chuẩn
- benchMEM
- benchRIB
- benchPEP
Các điểm chuẩn này là từ bộ điểm chuẩn Gromacs miễn phí của Tiến sĩ Kutzner và là một bộ tiêu chuẩn gồm các mô phỏng động lực học phân tử tạm thời. Mỗi phép đo điểm chuẩn có số lượng nguyên tử và thời lượng mô phỏng khác nhau. Các cấu hình có liên quan cho từng mô phỏng được trình bày trong bảng dưới đây.
Chỉ số / Điểm chuẩn | benchMEM | benchRIB | benchPEP |
Số lượng nguyên tử | 81.743 | 2.136.412 | 12.495.503 |
Kích thước hệ thống / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50 x 50 x 50 |
Time Step / fs | 2 | 4 | 2 |
Bán kính cắt / nm | 1 | 1 | 1.2 |
Khoảng cách lưới PME / nm | 0,12 | 0,135 | 0,16 |
Để chạy quy trình đo điểm chuẩn, bạn sẽ gửi một batch job Slurm. Theo mặc định, tập lệnh hàng loạt được cung cấp sẽ chạy phép đo điểm chuẩn benchRIB. Các bộ bài đầu vào xác định cấu hình cho các điểm chuẩn được cung cấp có trong hình ảnh máy ảo Gromacs trong /opt/share/gromacs. Ngoài ra, bạn có thể xem ví dụ về tập lệnh bash để chạy Gromacs trong /opt/share.
Đối với phần này, bạn phải SSH đến nút login của cụm
- Gửi một lô công việc bằng lệnh sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Thao tác này sẽ đưa công việc vào hàng đợi để thực thi và Slurm sẽ cung cấp một nút tính toán cho bạn. Khi chạy sinfo, bạn sẽ thấy một nút tính toán ở trạng thái alloc#, tức là nút tính toán được phân bổ cho công việc của bạn nhưng đang trong quá trình được cung cấp. Sau khi công việc của bạn đang chạy, nút sẽ được đặt ở trạng thái alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Chờ cho đến khi công việc hoàn tất. Điểm chuẩn mặc định (benchRIB) mô phỏng khoảng 8 triệu nguyên tử và được định cấu hình để chạy 5000 bước thời gian (với 4 bước thời gian/fs) và mất khoảng 6 phút để hoàn thành. Bạn có thể theo dõi trạng thái của lệnh này bằng lệnh sau:
watch squeue
Khi công việc của bạn đã được xử lý xong, bạn có thể thoát bằng cách nhấn tổ hợp phím ctrl-C.
- Khi hoàn tất, bạn sẽ thấy một thư mục có tên là
run/chứa đầu ra mô phỏng (trongrun/MEM) và một tệp nhật ký có tên làgromacs.outtrong thư mục hiện tại. Thư mụcrun/MEMchứa 2 tệpener.edrvàmd.log. Tệpener.edrlưu trữ năng lượng, nhiệt độ, áp suất, kích thước hộp, mật độ và virial của hệ thống ở định dạng nhị phân di động. Như phần mở rộng gợi ý, tệpmd.logchứa nhật ký cho mô phỏng Gromacs và bao gồm thông tin về hiệu suất mô phỏng, ngoài thông tin ghi nhật ký từ các trình giải hạt-hạt và PME. Nội dung của gromacs.out chứa đầu ra chuẩn từgmx mdrunsẽ có dạng tương tự như nội dung bên dưới.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Xin chúc mừng
Trong lớp học lập trình này, bạn đã tạo một cụm HPC tự động cấp tài nguyên bổ sung, gốc trên đám mây và chạy một mô phỏng động lực học phân tử được tăng tốc bằng GPU với Gromacs trên Google Cloud!
Dọn dẹp
Để tránh phát sinh phí cho tài khoản Google Cloud của bạn đối với các tài nguyên được dùng trong lớp học lập trình này, hãy làm như sau:
NÊN DÙNG: Xoá cụm HPC bằng Terraform
- Mở Cloud Shell rồi chuyển đến thư mục ví dụ
gromacs/tf/slurm
cd ~/rcc-apps/gromacs/tf/slurm
- Chạy lệnh make destroy để xoá tất cả tài nguyên.
make destroy
HOẶC, Xoá dự án (hiệu quả nhất và có tính huỷ diệt)
Cách dễ nhất để không bị tính phí là xoá dự án mà bạn đã tạo cho lớp học lập trình này.
Thận trọng: Việc xoá một dự án sẽ gây ra những tác động sau:
- Mọi nội dung trong dự án sẽ bị xoá. Nếu đã dùng một dự án hiện có cho lớp học lập trình này, thì khi xoá dự án đó, bạn cũng sẽ xoá mọi công việc khác mà bạn đã thực hiện trong dự án.
- Mã dự án tuỳ chỉnh sẽ bị mất. Khi tạo dự án này, có thể bạn đã tạo một mã dự án tuỳ chỉnh mà bạn muốn sử dụng trong tương lai. Để giữ lại những URL sử dụng mã dự án, chẳng hạn như URL appspot.com, hãy xoá các tài nguyên đã chọn trong dự án thay vì xoá toàn bộ dự án.
Nếu bạn dự định khám phá nhiều lớp học lập trình và hướng dẫn bắt đầu nhanh, thì việc sử dụng lại các dự án có thể giúp bạn tránh vượt quá hạn mức dự án.
- Trong Cloud Console, hãy chuyển đến trang Quản lý tài nguyên. Chuyển đến trang Quản lý tài nguyên
- Trong danh sách dự án, hãy chọn dự án mà bạn muốn xoá rồi nhấp vào biểu tượng Xoá
 .
. - Trong hộp thoại, hãy nhập mã dự án rồi nhấp vào Tắt để xoá dự án.