
1. Introduction
Dernière mise à jour : 2022-04-25
Objectif de cet atelier
Dans cet atelier de programmation, vous allez déployer un cluster de calcul haute performance (HPC) à autoscaling sur Google Cloud.Un déploiement Terraform crée ce cluster avec Gromacs installé via Spack. Le cluster sera géré avec le planificateur de tâches Slurm. Une fois le cluster créé, vous exécuterez les benchmarks benchMEM, benchPEP ou benchRIB.
Points abordés
- Déployer un cluster HPC compatible avec le planificateur de tâches Slurm
- Exécuter des simulations de dynamique moléculaire accélérées par GPU avec Gromacs sur Google Cloud à l'aide d'un job par lot Slurm
Prérequis
2. Configuration
Pour suivre cet atelier de programmation , vous devez activer les API Compute Engine et Cloud Build. Pour ce faire, ouvrez Cloud Shell et exécutez les commandes suivantes. N'oubliez pas de remplacer votre ID de projet par le
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Attention : Si vous prévoyez d'utiliser un SSH tiers (par exemple, OpenSSH) pour vous connecter à votre cluster, assurez-vous d'associer une clé SSH à votre profil Cloud Identity à l'aide de la connexion au système d'exploitation. Découvrez comment ajouter des clés SSH à votre profil Cloud Identity.
3. [FACULTATIF] Créer une image de VM GCP avec Gromacs
Pour cet atelier de programmation, nous vous fournissons une image précompilée, car le processus de compilation peut prendre jusqu'à deux heures pour installer Gromacs et toutes ses dépendances. Si vous souhaitez utiliser cette image précompilée pour gagner du temps, vous pouvez passer à la section suivante.
Lorsque vous exécutez des applications de recherche sur Google Cloud, vous disposez de nombreuses options pour installer et déployer votre application. Dans cette section de l'atelier de programmation, vous allez créer une image de machine virtuelle basée sur l'image de VM slurm-gcp (CentOS7). Lors du processus de création, le compilateur, toutes les dépendances de Gromacs et Gromacs seront installés.
Le pipeline Gromacs Cloud Build du dépôt RCC Apps encapsule les instructions nécessaires à l'installation de Gromacs. Le processus d'installation utilise Packer pour déployer une VM qui installe Spack, qui à son tour installe le compilateur GCC@9.2.0 et Gromacs@2021.2 avec l'accélération GPU activée.
- Ouvrez Cloud Shell sur GCP.
- Clonez le dépôt FluidNumerics/rcc-apps.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Créez l'image à l'aide de Google Cloud Build.
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Vous pouvez vérifier l'état de votre processus de compilation dans le tableau de bord Google Cloud Build.
Le processus de compilation peut prendre jusqu'à deux heures. Pour l'accélérer,vous pouvez envisager de modifier le schéma de votre fichier de configuration de compilation afin de changer le type de machine et d'améliorer les performances de compilation. Pour ce faire, vous pouvez utiliser la variable de compilation _MACHINE_TYPE. Exemple :
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Une fois la compilation terminée, une image de VM sera disponible dans votre projet Google Cloud. Vous pourrez l'utiliser pour déployer votre cluster.
4. Déployer un cluster HPC à autoscaling avec Terraform
Dans cette section, vous allez utiliser Terraform pour déployer un cluster HPC à autoscaling avec le planificateur de tâches Slurm installé. Ce cluster sera déployé avec des nœuds de calcul disposant chacun de huit processeurs virtuels et d'un GPU Nvidia® Tesla V100.
- Ouvrez Cloud Shell sur GCP.
- Clonez le dépôt FluidNumerics/rcc-apps.
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Accédez au répertoire gromacs terraform :
cd ~/rcc-apps/gromacs/tf/slurm
- Créez et examinez un plan Terraform. Définissez les variables d'environnement
GMX_NAME,GMX_PROJECTetGMX_ZONEpour spécifier le nom de votre cluster, votre projet GCP et la zone dans laquelle vous souhaitez effectuer le déploiement. Si vous n'êtes pas sûr, veuillez consulter la note ci-dessous.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Si vous avez créé votre propre image de VM dans la section précédente de cet atelier de programmation, vous devrez également définir la variable d'environnement GMX_IMAGE.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Créez le plan avec la commande make, qui exécutera
terraform init && terraform plan.
make plan
- Déployez le cluster. Le processus de configuration ne prend que quelques minutes, car Gromacs et ses dépendances sont préinstallés sur votre cluster.
make apply
- Connectez-vous en SSH au nœud login créé à l'étape précédente. Vous pouvez voir ce nœud à l'étape précédente (probablement appelé gromacs-login0). Pour ce faire, cliquez sur le bouton SSH à côté de la liste des instances de VM dans l'élément de menu de la console Compute Engine -> Instance de VM.
Option : Cette paire de commandes gcloud détermine le nom du nœud de connexion et s'y connecte en SSH :
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Une fois connecté au nœud de connexion, vérifiez la configuration de votre cluster en vous assurant que Gromacs est installé.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Vérifiez que
/opt/share/gromacscontient les éléments listés ci-dessous.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Exécuter le benchmark benchRIB
Gromacs est un logiciel de recherche utilisé pour simuler la dynamique moléculaire et calculer les structures moléculaires sous des contraintes de minimisation de l'énergie. Les benchmarks fournis dans l'image de VM pour cet atelier de programmation se concentrent sur la dynamique moléculaire, l'évolution des systèmes d'atomes.
Dans la dynamique moléculaire, les positions, les vitesses et les accélérations des atomes sont simulées à l'aide des lois du mouvement de Newton :
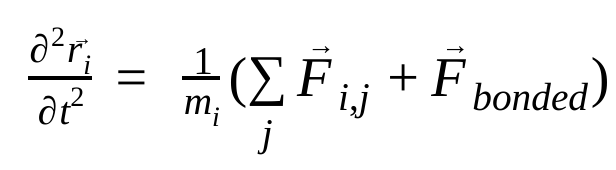
où 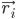 est la position de l'atome i, t est le temps,
est la position de l'atome i, t est le temps, 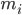 est la masse de l'atome i et
est la masse de l'atome i et 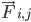 est la force non liée sur l'atome i due à l'atome j et
est la force non liée sur l'atome i due à l'atome j et 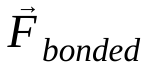 sont les forces dues aux interactions liées. Étant donné la température, la pression, les positions des atomes et les vitesses des atomes, les forces sont calculées et le système est intégré numériquement pour obtenir de nouvelles vitesses et positions des atomes. Ce processus est répété pour simuler la dynamique moléculaire pendant une période donnée.
sont les forces dues aux interactions liées. Étant donné la température, la pression, les positions des atomes et les vitesses des atomes, les forces sont calculées et le système est intégré numériquement pour obtenir de nouvelles vitesses et positions des atomes. Ce processus est répété pour simuler la dynamique moléculaire pendant une période donnée.
L'image Gromacs (celle que vous avez créée ou celle fournie) est fournie avec trois benchmarks.
- benchMEM
- benchRIB
- benchPEP
Ces benchmarks proviennent de l'ensemble de benchmarks Gromacs sans frais du Dr Kutzner et constituent un ensemble standard de simulations de dynamique moléculaire transitoire. Chaque benchmark varie en fonction du nombre d'atomes et de la durée de la simulation. Les configurations pertinentes pour chaque simulation sont indiquées dans le tableau ci-dessous.
Métrique / Benchmark | benchMEM | benchRIB | benchPEP |
Nombre d'atomes | 81 743 | 2 136 412 | 12 495 503 |
Taille du système / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50,0 x 50,0 x 50,0 |
Pas de temps / fs | 2 | 4 | 2 |
Rayons de coupure / nm | 1 | 1 | 1,2 |
Espacement de la grille PME / nm | 0,12 | 0,135 | 0,16 |
Pour exécuter le benchmark, vous devez envoyer un job par lot Slurm. Par défaut, le script par lot fourni exécute le benchmark benchRIB. Les decks d'entrée qui définissent les configurations des benchmarks fournis sont inclus dans l'image de VM Gromacs sous /opt/share/gromacs. De plus, un exemple de script bash pour exécuter Gromacs est disponible sous /opt/share.
Pour cette section, vous devez vous connecter en SSH au nœud login du cluster.
- Envoyez un job par lot à l'aide de la commande sbatch.
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Cela mettra le job en file d'attente pour l'exécution, et Slurm provisionnera un nœud de calcul pour vous. Lorsque vous exécutez sinfo, vous voyez qu'un nœud de calcul est à l'état alloc#, ce qui signifie qu'il est alloué à votre job, mais qu'il est en cours de provisionnement. Une fois votre job en cours d'exécution, le nœud sera défini sur l'état alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Attendez que la tâche soit terminée. Le benchmark par défaut (benchRIB) simule environ 8 millions d'atomes et est configuré pour exécuter 5 000 pas de temps (avec 4 pas de temps/fs) et prendre environ 6 minutes. Vous pouvez surveiller l'état de votre job à l'aide de la commande suivante :
watch squeue
Une fois votre job supprimé de la file d'attente, vous pouvez quitter avec Ctrl+C.
- Une fois le job terminé, vous devriez voir un répertoire appelé
run/contenant le résultat de la simulation (sousrun/MEM) et un fichier journal dans votre répertoire actuel appelégromacs.out. Le répertoirerun/MEMcontient deux fichiers :ener.edretmd.log. Le fichierener.edrstocke les énergies, la température, la pression, la taille de la boîte, la densité et les viriaux du système dans un format binaire portable. Comme son extension le suggère, le fichiermd.logcontient les journaux de la simulation Gromacs et inclut des informations sur les performances de la simulation, en plus des informations de journalisation des solveurs PME et particule-particule. Le contenu de gromacs.out contient la sortie standard degmx mdrunet doit ressembler à ce qui est présenté ci-dessous.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Félicitations
Dans cet atelier de programmation, vous avez créé un cluster HPC cloud natif à autoscaling et exécuté une simulation de dynamique moléculaire accélérée par GPU avec Gromacs sur Google Cloud.
Nettoyer
Pour éviter que les ressources utilisées dans cet atelier de programmation ne soient facturées sur votre compte Google Cloud, procédez comme suit :
RECOMMANDÉ : Supprimer le cluster HPC avec Terraform
- Ouvrez Cloud Shell et accédez au répertoire d'exemple
gromacs/tf/slurm.
cd ~/rcc-apps/gromacs/tf/slurm
- Exécutez make destroy pour supprimer toutes les ressources.
make destroy
OU, Supprimez le projet (méthode la plus efficace et la plus destructive).
Le moyen le plus simple d'empêcher la facturation est de supprimer le projet que vous avez créé pour cet atelier de programmation.
Attention : La suppression d'un projet aura les effets suivants :
- Tout le contenu du projet est supprimé. Si vous avez utilisé un projet existant pour cet atelier de programmation et que vous le supprimez, vous supprimerez également tout autre travail effectué dans le projet.
- Les ID de projets personnalisés sont perdus. Lorsque vous avez créé ce projet, vous avez peut-être créé un ID de projet personnalisé dont vous souhaitez vous servir par la suite. Pour conserver les URL qui utilisent l'ID de projet, telle qu'une URL appspot.com, supprimez les ressources sélectionnées dans le projet au lieu de supprimer l'ensemble du projet.
Si vous envisagez d'explorer plusieurs ateliers de programmation et guides de démarrage rapide, réutiliser des projets peut vous aider à ne pas dépasser les limites de quotas des projets.
- Dans la console Cloud, accédez à la page Gérer les ressources. Accéder à la page "Gérer les ressources"
- Dans la liste des projets, sélectionnez le projet que vous souhaitez supprimer, puis cliquez sur Supprimer
 .
. - Dans la boîte de dialogue, saisissez l'ID du projet, puis cliquez sur Arrêter pour supprimer le projet.