
1. Wprowadzenie
Ostatnia aktualizacja: 25.04.2022
Co utworzysz
W tym ćwiczeniu wdrożysz w Google Cloud klaster komputerów o dużej mocy obliczeniowej (HPC) z autoskalowaniem.Wdrożenie Terraform utworzy ten klaster z zainstalowanym programem Gromacs za pomocą Spack. Klaster będzie zarządzany za pomocą harmonogramu zadań Slurm. Po utworzeniu klastra uruchomisz testy porównawcze benchMEM, benchPEP lub benchRIB.
Czego się nauczysz
- Jak wdrożyć klaster HPC obsługujący harmonogram zadań Slurm
- Jak uruchamiać symulacje dynamiki molekularnej z akceleracją GPU za pomocą Gromacs w Google Cloud przy użyciu zadania wsadowego Slurm
Co będzie potrzebne
2. Konfiguracja
Aby ukończyć to ćwiczenie , musisz włączyć interfejsy Compute Engine API i Cloud Build API. Aby włączyć interfejsy Compute Engine API i Cloud Build API, otwórz Cloud Shell i uruchom te polecenia: Pamiętaj, aby zastąpić project-id w poniższym poleceniu znakiem
gcloud config set project <PROJECT_ID> gcloud services enable compute.googleapis.com gcloud services enable cloudbuild.googleapis.com
Ostrzeżenie: jeśli planujesz używać SSH innej firmy (np. OpenSSH) do łączenia się z klastrem, pamiętaj, aby dołączyć klucz SSH do profilu tożsamości w chmurze za pomocą OS Login. Więcej informacji o dodawaniu kluczy SSH do profilu tożsamości w chmurze
3. [OPCJONALNIE] Tworzenie obrazu maszyny wirtualnej GCP z Gromacs
Na potrzeby tego ćwiczenia udostępniliśmy gotowy obraz, ponieważ proces kompilacji może potrwać do 2 godzin, zanim zainstalujesz Gromacs i wszystkie jego zależności. Jeśli chcesz użyć tego gotowego obrazu, aby zaoszczędzić czas, możesz przejść do następnej sekcji.
Podczas uruchamiania aplikacji badawczych w Google Cloud masz wiele opcji instalowania i wdrażania aplikacji. W tej sekcji ćwiczenia w Codelabs utworzysz obraz maszyny wirtualnej, który będzie oparty na obrazie maszyny wirtualnej slurm-gcp (CentOS7). Podczas procesu tworzenia zostaną zainstalowane kompilator, wszystkie zależności Gromacs i Gromacs.
Potok Gromacs Cloud Build w repozytorium RCC Apps zawiera instrukcje niezbędne do zainstalowania Gromacs. Proces instalacji wykorzystuje Packera do wdrożenia maszyny wirtualnej, która instaluje Spacka, a ten z kolei instaluje kompilator GCC@9.2.0 i Gromacs@2021.2 z włączoną akceleracją GPU.
- Otwórz Cloud Shell w GCP.
- Klonowanie repozytorium FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- skompilować obraz za pomocą Google Cloud Build,
cd rcc-apps gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async
Stan procesu kompilacji możesz sprawdzić w panelu Google Cloud Build.
Proces kompilacji może potrwać do 2 godzin. Aby to przyspieszyć, możesz zmodyfikować schemat pliku konfiguracyjnego kompilacji i zmienić typ maszyny, aby zwiększyć wydajność kompilacji. Możesz to zrobić, korzystając ze zmiennej kompilacji _MACHINE_TYPE. Na przykład:
gcloud builds submit . --config=gromacs/cloudbuild.yaml --project=<PROJECT_ID> --async --substitutions=_MACHINE_TYPE=n2-standard-64
Po zakończeniu kompilacji w Twoim projekcie w chmurze Google Cloud będzie dostępny obraz maszyny wirtualnej, którego możesz użyć do wdrożenia klastra.
4. Wdrażanie klastra HPC z autoskalowaniem za pomocą Terraform
W tej sekcji użyjesz Terraform do wdrożenia klastra HPC z autoskalowaniem i zainstalowanym harmonogramem zadań Slurm. Ten klaster zostanie wdrożony z węzłami obliczeniowymi, z których każdy ma 8 vCPU i 1 GPU Nvidia® Tesla V100.
- Otwórz Cloud Shell w GCP.
- Klonowanie repozytorium FluidNumerics/rcc-apps
cd ~ git clone https://github.com/FluidNumerics/rcc-apps.git
- Przejdź do katalogu gromacs terraform:
cd ~/rcc-apps/gromacs/tf/slurm
- Utwórz i sprawdź plan Terraform. Ustaw zmienne środowiskowe
GMX_NAME,GMX_PROJECTiGMX_ZONE, aby określić nazwę klastra, projekt GCP i strefę, w której chcesz wdrożyć klaster. Jeśli nie masz pewności, zapoznaj się z poniższą uwagą.
export GMX_PROJECT=<PROJECT_ID> export GMX_ZONE=<ZONE> export GMX_NAME="gromacs"
- Jeśli w poprzedniej sekcji tego ćwiczenia utworzono własny obraz maszyny wirtualnej, musisz też ustawić zmienną środowiskową GMX_IMAGE.
export GMX_IMAGE="projects/${GMX_PROJECT}/global/images/gromacs-gcp-foss-latest"
- Utwórz plan za pomocą polecenia make, które uruchomi
terraform init && terraform plan.
make plan
- Wdróż klaster. Proces konfiguracji zajmuje tylko kilka minut, ponieważ Gromacs i jego zależności są fabrycznie zainstalowane w klastrze.
make apply
- Połącz się przez SSH z węzłem logowania utworzonym w poprzednim kroku. Ten węzeł możesz zobaczyć w poprzednim kroku (prawdopodobnie nazywa się gromacs-login0). Aby to zrobić, kliknij przycisk SSH obok listy instancji maszyn wirtualnych w pozycji menu konsoli Compute Engine –> Instancja maszyny wirtualnej.
Opcja: ta para poleceń gcloud określi nazwę węzła logowania i połączy się z nim przez SSH:
export CLUSTER_LOGIN_NODE=$(gcloud compute instances list --zones ${GMX_ZONE} --filter="name ~ .*login" --format="value(name)" | head -n1)
gcloud compute ssh ${CLUSTER_LOGIN_NODE} --zone ${GMX_ZONE}
- Po połączeniu się z węzłem logowania sprawdź, czy klaster jest skonfigurowany prawidłowo, i upewnij się, że zainstalowano Gromacs.
$ spack find gromacs ==> In environment /apps/spack-pkg-env ==> Root specs gromacs@2021.2 +cuda~mpi ==> 1 installed package -- linux-centos7-x86_64 / gcc@9.2.0 ----------------------------- gromacs@2021.2
- Sprawdź, czy
/opt/share/gromacszawiera wymienione poniżej elementy.
$ ls /opt/share/gromacs/ benchMEM.tpr benchPEP-h.tpr benchPEP.tpr benchRIB.tpr
5. Uruchomienie testu porównawczego benchRIB
Gromacs to oprogramowanie badawcze służące do symulowania dynamiki molekularnej i obliczania struktur molekularnych z uwzględnieniem ograniczeń związanych z minimalizacją energii. Testy porównawcze udostępnione w obrazie maszyny wirtualnej na potrzeby tego ćwiczenia skupiają się na dynamice molekularnej, czyli ewolucji układów atomów.
W dynamice molekularnej pozycje, prędkości i przyspieszenia atomów są symulowane przy użyciu praw ruchu Newtona :
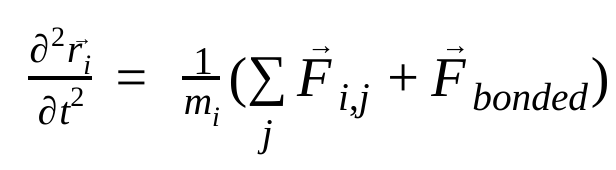
gdzie 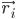 to pozycja atomu i, t to czas,
to pozycja atomu i, t to czas, 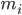 to masa atomu i,
to masa atomu i, 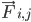 to siła oddziaływania niekierunkowego na atom i wywierana przez atom j, a
to siła oddziaływania niekierunkowego na atom i wywierana przez atom j, a 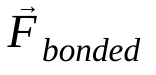 to siły wynikające z oddziaływań kierunkowych. Na podstawie temperatury, ciśnienia, pozycji i prędkości atomów obliczane są siły, a układ jest integrowany numerycznie w celu uzyskania nowych prędkości i pozycji atomów. Ten proces jest powtarzany, aby zasymulować dynamikę molekularną w danym okresie.
to siły wynikające z oddziaływań kierunkowych. Na podstawie temperatury, ciśnienia, pozycji i prędkości atomów obliczane są siły, a układ jest integrowany numerycznie w celu uzyskania nowych prędkości i pozycji atomów. Ten proces jest powtarzany, aby zasymulować dynamikę molekularną w danym okresie.
Obraz Gromacs (utworzony przez Ciebie lub udostępniony) zawiera 3 testy porównawcze.
- benchMEM
- benchRIB
- benchPEP
Te testy porównawcze pochodzą z zestawu bezpłatnych testów porównawczych Gromacs dr. Kutznera i stanowią standardowy zestaw przejściowych symulacji dynamiki molekularnej. Każdy test porównawczy różni się liczbą atomów i czasem trwania symulacji. Odpowiednie konfiguracje dla każdej symulacji znajdziesz w tabeli poniżej.
Wskaźnik / test porównawczy | benchMEM | benchRIB | benchPEP |
Liczba atomów | 81 743 | 2 136 412 | 12 495 503 |
Rozmiar systemu / nm | 10,8 x 10,2 x 9,6 | 31,2 x 31,2 x 31,2 | 50,0 x 50,0 x 50,0 |
Krok czasowy / fs | 2 | 4 | 2 |
Promienie odcięcia / nm | 1 | 1 | 1,2 |
Odstępy siatki PME / nm | 0,12 | 0.135 | 0,16 |
Aby uruchomić test porównawczy, prześlij zadanie wsadowe Slurm. Domyślnie podany skrypt wsadowy uruchamia test porównawczy benchRIB. Pliki wejściowe, które określają konfiguracje podanych testów porównawczych, są zawarte w obrazie maszyny wirtualnej Gromacs w folderze /opt/share/gromacs. Dodatkowo w sekcji /opt/share znajdziesz przykładowy skrypt bash do uruchamiania Gromacs.
W tej sekcji musisz połączyć się przez SSH z węzłem logowania klastra.
- Przesyłanie zadania wsadowego za pomocą polecenia sbatch
$ sbatch --ntasks=1 --cpus-per-task=8 --gres=gpu:1 --out=gromacs.out /opt/share/gromacs_bench.sh
Spowoduje to umieszczenie zadania w kolejce do wykonania, a Slurm udostępni Ci węzeł obliczeniowy. Gdy uruchomisz polecenie sinfo, zobaczysz, że węzeł obliczeniowy jest w stanie alloc#, co oznacza, że jest on przydzielony do Twojego zadania, ale jest w trakcie udostępniania. Gdy zadanie będzie uruchomione, węzeł zostanie ustawiony w stanie alloc.
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc# gromacs-compute-0-0
$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
2 gromacs gromacs_ joe R 0:02 1 gromacs-compute-0-0
$ sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
gromacs* up infinite 1 alloc gromacs-compute-0-0
Poczekaj na zakończenie zadania. Domyślny test porównawczy (benchRIB) symuluje około 8 milionów atomów i jest skonfigurowany tak, aby wykonać 5000 kroków czasowych (4 kroki czasowe na femtosekundę) i zająć około 6 minut. Stan zadania możesz sprawdzić za pomocą tego polecenia:
watch squeue
Gdy zadanie zniknie z kolejki, możesz wyjść, naciskając Ctrl+C.
- Po zakończeniu zadania w bieżącym katalogu powinien pojawić się katalog o nazwie
run/, który zawiera dane wyjściowe symulacji (w katalogurun/MEM) i plik dziennika o nazwiegromacs.out. Katalogrun/MEMzawiera 2 pliki:ener.edrimd.log. Plikener.edrprzechowuje energie, temperaturę, ciśnienie, rozmiar pudełka, gęstość i wartości wirialne systemu w przenośnym formacie binarnym. Jak sugeruje rozszerzenie, plikmd.logzawiera logi symulacji Gromacs i informacje o jej wydajności, a także informacje z rozwiązań particle-particle i PME. Zawartość pliku gromacs.out zawierającego standardowe dane wyjściowe poleceniagmx mdrunpowinna wyglądać podobnie do tego, co widać poniżej.
$ tail -n9 gromacs.out
step 5000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 3085.342 385.683 800.0
(ns/day) (hour/ns)
Performance: 4.481 5.356
GROMACS reminds you: "I never thought of stopping, and I just hated sleeping. I can't imagine having a better life." (Barbara McClintock)
OK
6. Gratulacje
W tym ćwiczeniu utworzyliśmy klaster HPC z autoskalowaniem natywny dla chmury i przeprowadziliśmy symulację dynamiki molekularnej z akceleracją GPU za pomocą programu Gromacs w Google Cloud.
Czyszczę dane
Aby uniknąć obciążenia konta Google Cloud opłatami za zasoby użyte w tym ćwiczeniu:
ZALECANE: usuń klaster HPC za pomocą Terraform
- Otwórz Cloud Shell i przejdź do katalogu z przykładem
gromacs/tf/slurm.
cd ~/rcc-apps/gromacs/tf/slurm
- Uruchom polecenie make destroy, aby usunąć wszystkie zasoby.
make destroy
LUB usuń projekt (najskuteczniejsze i najbardziej destrukcyjne rozwiązanie).
Najprostszym sposobem na uniknięcie płatności jest usunięcie projektu utworzonego na potrzeby tego ćwiczenia w Codelabs.
Ostrzeżenie: usunięcie projektu spowoduje, że:
- Wszystkie dane projektu zostaną usunięte. Jeśli w tym ćwiczeniu w Codelabs używasz istniejącego projektu, po jego usunięciu usuniesz też wszystkie inne wykonane w nim zadania.
- Niestandardowe identyfikatory projektu zostaną utracone. Podczas tworzenia tego projektu możesz utworzyć niestandardowy identyfikator projektu, którego chcesz używać w przyszłości. Aby zachować adresy URL, które używają identyfikatora projektu, np. adres URL appspot.com, usuń wybrane zasoby w projekcie zamiast usuwać cały projekt.
Jeśli zamierzasz skorzystać z innych ćwiczeń z programowania i krótkich wprowadzeń, ponowne wykorzystanie tych samych projektów może pomóc Ci uniknąć przekroczenia Iimitów związanych z projektami.
- W Cloud Console otwórz stronę Zarządzanie zasobami. Otwórz stronę Zarządzanie zasobami
- Z listy projektów wybierz projekt do usunięcia, a potem kliknij Usuń
 .
. - W oknie wpisz identyfikator projektu i kliknij Wyłącz, aby usunąć projekt.